
ATP7B基因突变与肝豆状核变性发病关系的探讨
2015-03-20 09:48余元勋鲍远程王迎新李建平
安徽医科大学学报 2015年12期
徐 彬,余元勋,鲍远程,王迎新,刘 萍,李建平,朱 霖
ATP7B基因突变与肝豆状核变性发病关系的探讨
徐 彬1,余元勋1,鲍远程2,王迎新1,刘 萍1,李建平1,朱 霖1
目的对中国人肝豆状核变性(WD)患者ATP7B全基因测序,分析其突变及与WD的关系,并探讨ATP7B基因复合突变在WD发病中的意义。方法对临床确诊的WD患者84例及20例健康者采集口腔黏膜细胞,提取基因组DNA为模板,应用聚合酶链反应对ATP7B全部外显子5′端→3′端扩增;运用DNA直接测序法检测突变,并分析ATP7B基因突变临床意义及与预后的关系。结果在84例患者中,ATP7B基因突变率为82.14%,还发现一些国内较少报道的ATP7B基因复合突变,可能与WD相关。结论ATP7B基因突变有较大的异质性,对该基因突变的检测,能发现有相关ATP7B基因突变的WD患者。
肝豆状核变性;外显子;基因突变;聚合酶链反应
肝豆状核变性(Wilson disease,WD)是一种常染色体单基因隐性遗传的铜代谢障碍疾病,大多在5~35岁起病,人群发病率为1/30 000,携带者概率为1/90[1-2];一般肝病出现较早,神经系统症状常在青春期后出现[3]。WD致病相关的P-型铜转运酶(ATP7B)基因定位于13ql4.3,含21个外显子、20个内含子,编码ATP7B[4],后者参与铜跨膜转运[5-6]。ATP7B基因突变能导致ATP7B改变、WD引起铜蓝蛋白合成减少、铜离子排出受阻,铜在肝、脑、角膜等组织沉积,导致多脏器损害[7]、肝硬化、神经/精神症状、角膜K-F环、尿铜增加等。ATP7B基因突变常分布在各外显子,迄今已发现至少651种突变,突变形式有错义/无义突变(占63%)、小片段缺失(占17%)、小片段插入(占8%)、剪接突变(占9%)、大片段缺失(占2%)、同义突变(占2%)[8]等;国内以前较少报道复合突变。作者对84例WD患者ATP7B基因进行全外显子+5′端→+3′端的测序,旨在探讨ATP7B基因的突变的异质性及与WD发病的关系。
1 材料与方法
1.1 病例资料选取安徽中医药大学第一附属医院2009年5月~2014年7月收治的84例中国汉族WD患者,诊断标准参照中华医学会WD诊断指南[9]及美国的AASLD的WD诊断及治疗指南[10];其中男55例,女29例;年龄6~56(20.9±3.6)岁;其中49例为先证者,其余35例来自于13个患者家庭。20例中国汉族健康输血员,男14例,女6例;年龄21~32(27.7±2.8)岁,作为对照组。所采样本对象有临床资料及知情同意书。本研究旨在对临床上确诊为WD或疑似WD的患者家系成员,采用分子生物学技术进行遗传学确诊,能确定ATP7B基因突变型、SNPs。
1.2 方法
1.2.1 生化测定 WD组及对照组的WD相关生化测定16项由安徽中医药大学第一附属医院按常规完成,并应用软件对两组化验结果进行统计学分析。
1.2.2 基因诊断 本研究参考方法[11]进行基因诊断,简述如下:对所有明确诊断的WD患者、参与研究的家属、对照者,取口腔黏膜细胞,制备基因组DNA溶液,凝胶电泳为一条带,达电泳纯;基因组DNA溶液经紫外分光光度计检测,测量光密度(optical density,OD)值,OD260/OD280为1.8,纯度合格;基因组DNA溶液浓度为1 μg/μl。引物设计参考美国国立生物技术信息中心的信息,根据参考文献[11]并利用Premier 5.0软件设计外显子5′端→3′端的21对PCR扩增引物,由上海生工生物公司合成。反应体系和扩增条件及产物测序按文献[11]进行。测序PCR的反应产物经荧光全自动DNA正向测序及反向测序,分析ATP7B基因各外显子5′端→3′端的突变,对后者中有疑问者,均经PCR克隆测序后再确认。测序仪:ABI3730型;分析软件:SeqMan、BioEdit。WD患者和健康者ATP7B基因测序结果与GenBank中正常者ATP7B基因相应序列比对,分析突变情况。
1.3 统计学处理生化检测所得数据以Excel建库,采用SPSS 19.0软件进行分析,计量资料以表示。数据做正态性W检验和方差齐性Levene检验,采用随机区组设计的方差分析,比较组间差异。ATP7B基因的所有外显子5′端→3′端,DNA的测序结果,与由GenBank的正常者ATP7B基因相应序列进行比对,分析突变情况。
2 结果
2.1 生化检测结果84例WD患者及20例健康者的生化检测结果见表1。84例WD患者血清铜离子、铜氧化酶、铜蓝蛋白(ceruloplasmin,CP)的水平明显低于20例健康者,差异有统计学意义(P<0.01),其中ATP7B基因突变者的血清铜离子、铜氧化酶、CP水平均明显降低。
表1 84例WD患者及20例健康者生化检测结果()

表1 84例WD患者及20例健康者生化检测结果()
项目WD组对照组P值年龄(岁)19.240±3.57721.700±2.680-年限(年)4.560±4.2480-白细胞(×109/L)4.462±1.5726.141±1.425<0.05红细胞(×1012/L)3.862±0.5485.583±0.639<0.05血红蛋白(g/L)120.737±21.536 129.612±15.132-血小板(×109/L)93.526±37.216179.630±47.720<0.05 ALT(IU/L)78.371±49.23728.710±8.328<0.05 AST(IU/L)69.710±47.31033.260±9.851<0.05总胆汁酸(μmol/L)38.826±31.9586.539±3.253<0.05总胆红素(μmol/L)28.275±20.77214.264±5.349<0.05直接胆红素(μmol/L)18.352±13.7755.217±1.391<0.05血清白蛋白比(g/L)39.172±7.72843.629±4.612<0.05白/球蛋白(mmol/L)1.856±0.4441.827±0.317-尿素氮(mmol/L)4.824±1.3704.731±1.297-肌酐(μmol/L)63.032±18.94178.235±20.221<0.05血清铜(μmol/L)8.993±5.87117.147±3.352<0.01铜氧化酶(活性单位/L)0.074±0.0670.421±0.113<0.01 CP(g/L)0.086±0.0300.321±0.031<0.01
2.2 WD患者ATP7B的基因突变测序本次研究通过对84例WD患者的ATP7B基因的全部21个外显子5′端→3′端进行直接DNA测序,显示WD患者ATP7B基因突变率较高,种类较多,共检出23例纯合子突变(18例Arg778Leu纯合子突变和5例Arg919Gly纯合子突变,占总突变的27.38%),36例单纯杂合子突变(占总突变的42.86%),10例复合突变(占总突变的11.90%),15例未检出突变(占总突变的17.86%)。84例WD患者168个ATP7B等位基因中的突变总检出率为82.14%,共检测出致病突变12种,其中有5种复合突变,突变位点分布于全部外显子,表现在8、10、12、13、16、21外显子,其中6种单纯突变为:Arg969Glm、Ser975Tyr、Arg778Leu、Gly988Val、Pro992Leu、Ala1018Val,5种复合突变的具体信息见表2。在20例健康对照者中没有ATP7B基因突变。通过在PubMed上进行文献检索,以及在http://www.wilsondisease.med.ualberta.Ca/database.asp和http://www.hgmd.cf.ac.uk/ac/validate.phP进行查询,结果显示5种复合突变国内报道较少,这5种ATP7B基因复合突变的相应的测序图见图1~5。
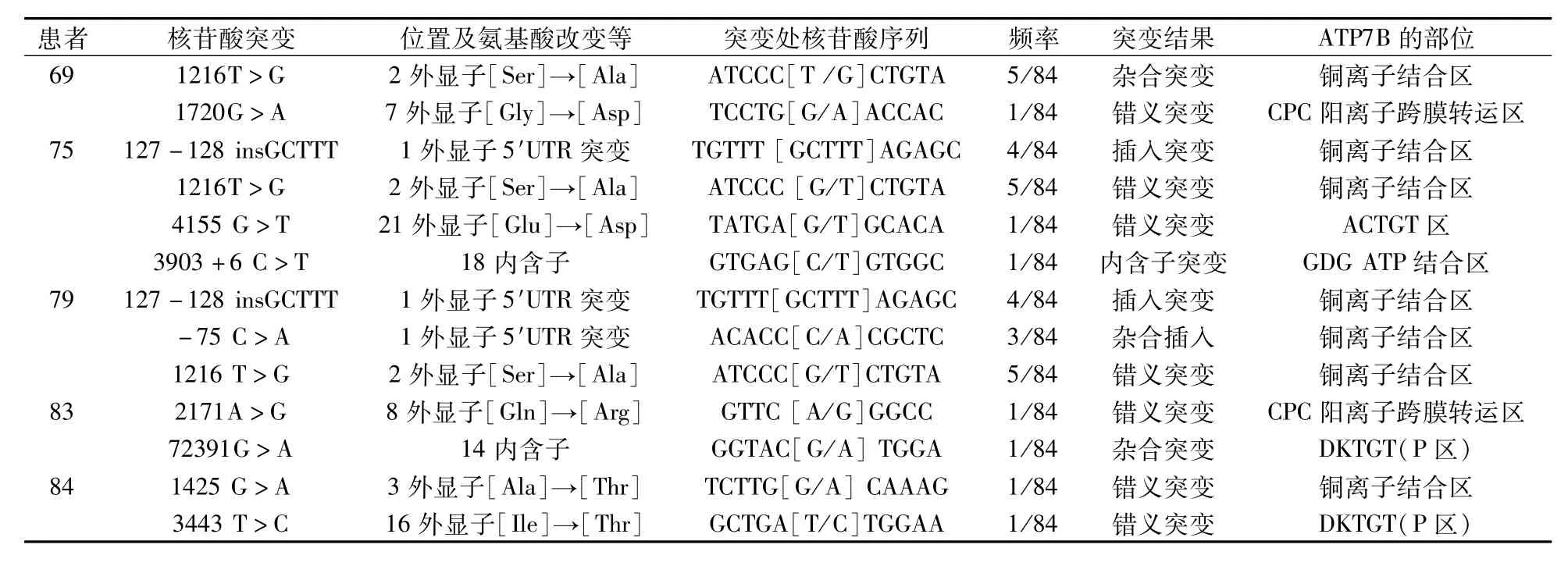
表2 WD患者ATP7B的基因突变
3 讨论
WD患者中约42%以肝脏损伤为首发表现,约44%以基底神经节及大脑其他区域铜沉积而呈现神经、精神系统症状为首发表现[3]。本研究中ATP7B基因突变患者血清铜离子、铜氧化酶、CP的水平明显低于正常对照组,差异有统计学意义,表明WD患者ATP7B基因突变失活后,肝排铜减少,使患者出现WD相关血清生化检验(+),与WD临床发病呈正相关性。
ATP7B是铜离子转运P型ATP酶,含1 411个氨基酸残基,分子量约150 ku,在细胞外铜离子水平正常时,ATP7B位于细胞核周围;在细胞外铜离子水平升高并大量进入细胞时,ATP7B转移到细胞质,结合铜离子后,促进ATP结合区结合ATP,使ATP水解释放能量,使天冬氨酸激酶磷酸化区(P区)磷酸化,然后ATP7B转位到细胞膜,经毛细胆管处排出铜离子。
当ATP7B发生突变时,导致其功能部分或全部丧失,使铜离子经胆汁排出减少,同时CP合成减少,使血CP减少,血中白蛋白疏松结合的铜离子增加,易沉积到中枢神经系统、肝脏、肾脏、角膜等处,造成相应脏器的形态结构破坏、功能改变而致病。
本研究表明,Arg969Glm、Ser975Tyr、Arg778Leu、Gly988Val、Pro992Leu、Ala1018Val等单纯突变,在WD患者阳性率相对较高(占总突变的42.86%),10例复合突变(占总突变的11.9%);由于对ATP7B全基因外显子扫描,本文突变总检出率较高为82.14%,5种复合突变少见报道,突变位点分布于全部外显子,与其他研究[12]结果相一致;如再对内含子扫描,突变检出率可进一步提高[13]。说明ATP7B基因突变可引发单基因遗传WD,可能是WD发病的主要原因。
WD患者ATP7B基因的复合突变形式多样,69号患者有外显子2、7复合突变,第2外显子突变出现在2铜离子结合区,使ATP7B不能结合铜离子、不能活化,排出铜离子减少;第7外显子突变出现在CPC阳离子跨膜转运区,使ATP7B一直停留在核周高尔基体外侧网络,不能到细胞膜排出铜离子。75号患者有外显子1、2、21及18内含子区突变,第1、2外显子突变出现在1、2铜离子结合区,使ATP7B不能结合铜离子、不能活化,排出铜离子减少;第21外显子突变出现在C末端区,阻碍调节ATP7B功能;第18内含子区纯合突变,使ATP7B表达水平降低;结果可引起铜转运停滞,发生疾病。79号患者有外显子1、2突变,第1、2外显子突变出现在1、2铜离子结合区,使ATP7B不能结合铜离子、不能活化,排出铜离子减少。83号患者有外显子8、14内含子区突变,第8外显子突变出现在CPC阳离子跨膜转运区,使ATP7B一直停留在内质网,易被泛素蛋白酶体降解,不能到细胞膜排出铜离子;14内含子区存在杂合突变,使ATP7B表达水平降低,可引起铜转运停滞。84号患者有外显子3、16突变,第3外显子突变出现在3铜离子结合区,使ATP7B不能结合铜离子、不能活化,排出铜离子减少;第16外显子突变,使ATP7B不能形成ATP7B-酰基磷酸盐,使ATP不能供应能量,使ATP7B不能移位转运铜离子。
对于临床检查和实验室检测后仍很难确诊的WD患者,通过对ATP7B基因进行分子遗传学检测,明确致病的等位基因来确诊非常有用。随着研究的深入,WD的检出率最高可达100%[12]。文献报道,ATP7B基因复合突变患者一般比单纯突变患者的临床症状常更重,Maier-Dobersberger et al[14]对ATP7B基因突变位点研究后认为,复合突变常比单一突变发病更早。
[1] Dedoussis G V,Genechel J,Sialvera T E,et al.Wilson disease:high prevalence in a mountainous area of Crete[J].Ann Hum Genet,2005,69(Pt 3):268-74.
[2] Burkhead J L,Gray L W,Lutsenko S.Systematic biology approach to Wilson disease[J].Biometals,2011,24(3):455-66.
[3] Chan H W,Liu T,Verdile Q,et al.Copper induces apoptosis of neuroblastoma cells via post-translational regulation of the expression of Bcl-2 family proteins and the tx mouse is a better model of hepatic than brain Cu toxicity[J].Int J Clin Exp Med,2008,1(1):76-88.
[4] Gitlin J D.Wilson disease[J].Gastroenterology,2003,125(6):1868-77.
[5] Nicastro E,Loudianos G,Zancan L,et al.Genotype-phenotype correlation in Italian children with Wilson disease[J].J Hepatol,2009,50(3):555-61.
[6] Telianidis J,Hung Y H,Materia S,et al.Role of the P-Type ATPases,ATP7A and ATP7B in brain copper homeostasis[J].Front Aging Neurosci,2013,5:44.
[7] Lee J Y,Kim Y H,Kim T W,et al.New novel mutation of the ATP7B gene in a family with Wilson disease[J].J Neurol Sci,2012,313(1-2):129-31.
[8] Seo J K.Diagnosis of Wilson disease in young children:molecular genetic testing and a paradigm shift from the laboratory diagnosis[J].Pediatr Gastroenterol Hepatol Nutr,2012,15(4):197-209.
[9] 中华医学会神经病学分会帕金森病及运动障碍学组,中华医学会神经病学分会神经遗传病学组.肝豆状核变性的诊断与治疗指南[J].中华神经科杂志,2008,41(8):566-9.
[10]Roberts E A,Schilsky M L,AASLD.Diagnosis and treatment of Wilson disease:an update[J].Hepatology,2008,47(6):2089 -111.
[11]徐 彬,余元勋,鲍远程,等.肝豆状核变性中ATP7B基因复合突变的研究[J].第二军医大学学报,2014,35(11):1209-14.
[12]Moller L B,Horn N,Jeppesen T D,et al.Clinical presentation and mutations in Danish patients with Wilson disease[J].Eur J Hum Genet,2011,19(9):935-41.
[13]黄 丽,李洵桦,梁秀龄,等.各种辅助检查对肝豆状核变性的诊断价值[J].临床神经病学杂志,2006,19(1):8-11.
[14]Maier-Dobersberger T,Ferenci P,Polli C,et al.Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction[J].Ann Intern Med,1997,127(1):21-6.
Research for the relationship of ATP7B gene mutations and WD
Xu Bin1,Yu Yuanxun1,Bao Yuancheng2,et al
(1Anhui Medical College,Anhui Provice Medical Genetics Center,Hefei 230061;
2Dept of Neurology,The First Affiliated Hospital of Anhui Traditional Chinese Medicine University,Hefei 230031)
ObjectiveTo sequence the WD ATP7B gene,analyze the relationship of the mutations and WD,find in Chinese WD patients the ATP7B gene complicated mutation meaning.Method Geting genomic DNA from clinically diagnosed 84 cases of WD patients and 20 cases of the health people,amplifying all exons 5′end→+3′end of ATP7B gene by PCR.PCR products and mutations were analyzed by DNA direct sequencing and analyzing the clinical meaning and the relationship with the mutations.ResultsWe found in the 84 cases the ATP7B gene mutation rate was 82.14%and found some complicated mutations that were rarely reported in China and had relatioship with WD.ConclusionATP7B gene mutations have the heterogenecity and the mutation checking can find related ATP7B gene mutation carried WD patients.
Wilson disease;exons;gene mutations;PCR
R 742.4
A
1000-1492(2015)12-1762-05
时间:2015-11-18 10:12:34
http://www.cnki.net/KCMS/detail/34.1065.R.20151118.1012.024.html
2015-10-09接收
安徽高等学校省级自然科学研究项目(编号:KJ2014A128)
1安徽医学高等专科学校安徽省遗传医学中心“原安徽医
科大学遗传室”,合肥 230061
2安徽中医药大学第一附属医院神经内科,合肥 230031
徐 彬,男,副研究员,责任作者,E-mail:xb.1030@163.com
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
中国生殖健康(2020年4期)2021-01-18
山地农业生物学报(2020年2期)2020-11-09
现代农业科技(2020年15期)2020-08-16
环球时报(2019-04-03)2019-04-03
生物学教学(2018年12期)2018-08-15
湖北农业科学(2014年11期)2014-09-10
