
邻苯二酚烷基化反应机理的密度泛函研究❋
2015-03-18 08:21:58吴晓静夏树伟于良民陈国博中国海洋大学1海洋化学理论与工程技术教育部重点实验室化学化工学院山东青岛266100
中国海洋大学学报(自然科学版) 2015年8期
吴晓静, 夏树伟❋❋, 于良民, 陈国博(中国海洋大学1.海洋化学理论与工程技术教育部重点实验室; 2.化学化工学院,山东 青岛 266100)
邻苯二酚烷基化反应机理的密度泛函研究❋
吴晓静1,2, 夏树伟1,2❋❋, 于良民1,2, 陈国博1,2
(中国海洋大学1.海洋化学理论与工程技术教育部重点实验室; 2.化学化工学院,山东 青岛 266100)
采用密度泛函理论(DFT)B3LYP方法,在6-311G(d,p)基组下研究邻苯二酚与N-羟甲基丙烯酰胺的烷基化反应机理,对反应物、过渡态、中间体和产物进行了结构全优化和确认,通过HOMO轨道百分组成预测芳环上的反应活性位点。结果表明:邻苯二酚与N-羟甲基丙烯酰胺烷基化反应机理分四步,活化能分别是61.07、39.69、44.31和31.51J/mol。一取代反应势垒为61.07kJ/mol;二取代反应势垒为44.31kJ/mol。表明二取代反应较一取代更容易发生。另外,一取代产物芳环上活性位点碳原子所带负电荷(-0.806)高于反应物邻苯二酚芳环上的碳原子(-0.124),有利于亲电试剂进攻,说明反应更易发生二取代,这与实验结果一致。
邻苯二酚;反应机理;Friedel-Crafts烷基化反应;密度泛函研究
从辣椒、胡椒等辛辣植物中提取的辣素具有驱虫、抗菌、防污、抗肿瘤和镇痛消炎等作用,广泛应用于农药、医药、轻工等方面[1-3]。然而从植物中提取辣素产量低、成本高,不能满足社会需要。人工合成廉价易得的辣素及其类似物,可用于抗菌防污等[4-7]。
邻苯二酚和N-羟甲基丙烯酰胺经烷基化反应,生成辣素类似物N-(3、4-二羟基-6-丙烯酰胺甲基苄基)丙烯酰胺,反应式如图1所示。实验中发现,在较佳反应条件下,改变初始反应物配比分别为1∶1、5∶1、10∶1时,该反应均只得到二取代产物,而没有单取代产物生成。
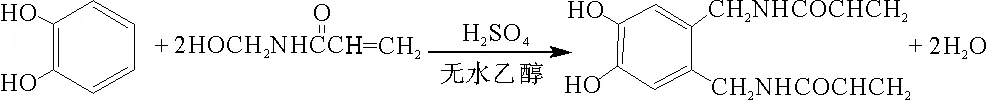
图1 邻苯二酚和N-羟甲基丙烯酰胺反应式
Fig.1 Reaction of pyrocatechol and N-methylolacrylamide
邻苯二酚与N-羟甲基丙烯酰胺的反应为芳香族亲电取代反应,人们对芳香族亲电反应机理进行了实验和理论研究[8-11]。齐国鹏[12-13]、孙学文[14]等研究了催化苯衍生物与烯烃的亲电取代反应,利用同位素取代示踪法推导了反应机理,认为亲电试剂对芳环上富电子活性位进攻,经过一个正离子中间体,生成烷基化产物。Namba等[15-18]研究了苯酚与醇类的烷基化反应,苯酚易产生稳定的苯氧负离子,在弱酸或碱性条件下易发生O-烷基化,较强酸可与苯氧负离子相互作用,有效抑制O-烷基化,反应活性与催化剂酸性正相关。徐亚荣等[19]采用从头算方法研究了不同的甲基取代的噻吩衍生物的烷基化反应机理,认为是分步的正离子中间体机理。聂小娃等[20-22]采用DFT方研究法了分子筛及离子液体酸催化芳香化合物和醇的烷基化反应机理,包括生成烷基化中间体和中间体去质子化两个过程。然而,这些关于芳香族烷基化反应机理的研究均只针对单取代反应进行的,且对脱质子过程的描述模糊不清。本文从静态和动态两方面研究了邻苯二酚与N-羟甲基丙烯酰胺的反应机理,获得了过渡态、中间体的分子结构、活化能、反应途径及电子结构等参量,发现水分子协助脱除质子,并合理地解释了该反应二取代优于单取代的实验现象。
1 计算模型和方法
用密度泛函理论,在B3LYP/6-311G(d,p)下对所有的反应物、中间体、过渡态和产物的几何结构进行全优化,并进行频率分析确认。用STQN/QST3方法寻找反应过程中的过渡态,并对过渡态进行内禀反应坐标(IRC)解析,确定过渡态的正确性。在同一理论水平上计算其能量,并进行零点能校正。对反应中涉及的所有分子结构模型,均在气相优化结构的基础上采用IEFPCM极性溶剂模型来模拟乙醇溶剂环境。所有计算由Gaussian09完成。
2 结果与讨论
2.1 反应活性位点
芳香亲电反应具有明显的区域选择性,亲电试剂优先进攻电子丰富的芳环位点。付蓉[23]对芳香亲电取代反应的活性位点进行预测,比较了14种预测方法的可靠性,其中HOMO轨道成分能够较好地预测双取代苯体系的活性位点。本文分析的HOMO成分是基于自然键轨道(NBO)的方法,邻苯二酚、一取代物P1及产物A的HOMO轨道组成如表1所示。
表1 各构型的HOMO轨道组成
Table 1 Atomic orbitals contribution to some frontier HOMO /%
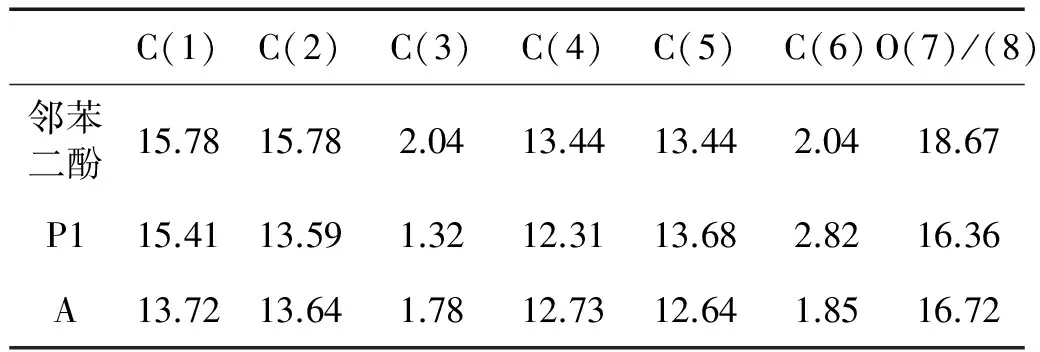
由表1数据可知,邻苯二酚、一取代物P1及产物A的HOMO轨道主要分布在苯环碳原子及酚羟基氧原子上。由于酚羟基氧原子的非键pz轨道与苯环的π成键轨道之间存在共振作用,使酚羟基氧原子上具有较高的电子密度,HOMO轨道的18.67%,易发生O-烷基化反应。本实验的强酸条件能够较好的抑制O-烷基化,主要反应为C-烷基化。邻苯二酚中C(4)、C(5) HOMO轨道百分组成为13.44%,高于C(3)、C(6)的2.04%,是该反应条件下最可能的反应位点。对邻苯二酚的一取代物P1,可能的位点中C(5)位对HOMO轨道的贡献最大,是最可能发生再次取代的位置。反应活性预测结果与最终产物一致。在亲电取代反应中,通过前线轨道计算反应活性位点,可以简单预测可能的反应产物。
2.2 前线分子轨道能级
单取代反应过程中邻苯二酚、[CH2NHCOCHCH2]+、一取代产物P1的前线分子轨道能级见表2,亲电试剂[CH2NHCOCHCH2]+的LUMO轨道与邻苯二酚HOMO轨道能级差为1.739eV,与P1的HOMO轨道能级差为1.614eV,可见[CH2NHCOCHCH2]+与P1的前线轨道能级更接近,因此两者之间更易发生反应。
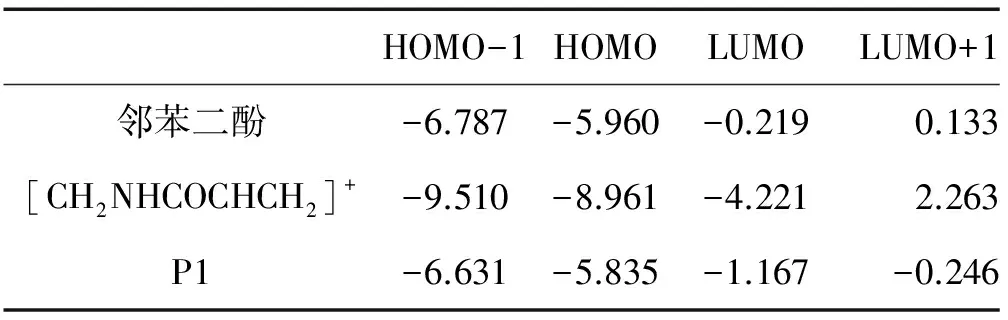
表2 几种分子的前线分子轨道能级Table 2 Frontier molecular orbital energy levels /eV
2.3 反应途径
在强酸催化下,邻苯二酚与N-羟甲基丙烯酰胺发生烷基化反应,在该条件下应为正离子机理,反应途径如图2所示。
N-羟甲基丙烯酰胺端位羟基氧原子上的孤对电子和硫酸的氢离子相互作用诱导C—O键异裂,形成碳正离子。碳正离子作为亲电试剂,进攻邻苯二酚π体系,经过渡态TS1,形成中间体M1。随着C—C键的形成苯环C原子由sp2→sp3,中间体M1的H与H2O中的O键合,经过渡态TS2,H以H3O+形成脱离,体系恢复芳香性,形成一烷基化产物P1。二取代过程与单取代类似,此时一取代产物P1为反应物,其负电性C原子被亲电试剂进攻,形成中间体M2。M2脱去质子生成产物P2,即最终产物(A)。亲电进攻(1)、(3)是速控步,脱质子(2)、(4)形成芳香性产物,为放热过程,是整个反应的驱动力[24]。

图2 邻苯二酚与N-羟甲基丙烯酰胺反应途径示意图Fig.2 Reaction path of alkylation of pyrocatechol with N-methylolacrylamide
2.4 反应物、中间体、过渡态、产物的优化构型
对反应过程中的所有反应物、过渡态、中间体和产物的结构进行全优化,其结构图见图3。
邻苯二酚和N-羟甲基丙烯酰胺的烷基化属于芳香族亲电取代反应,[CH2NHCOCHCH2]+正离子为亲电试剂,进攻苯环上C原子。虽受邻、对位活化基团酚羟基的影响,邻苯二酚中苯环碳原子间键长差异不大,芳环共轭性较好。邻苯二酚中C(3)、C(4)、C(5)、C(6)均带负电荷,对位C(4)(或C(5))所带负电荷略高(-0.124),且位阻较小,亲电试剂优先进攻。随着亲电试剂端位碳原子C(9)与邻苯二酚C(4)的逐渐接近,经过渡态TS1形成中间体M1。过渡态TS1中C(4)-C(9)距离为2.115Å,此时C(4)-H(7)中的H(7)原子略翘起,与芳环平面的夹角为12.296°。中间体M1中C(4)-C(9)(1.591Å)单键形成。C(3)-C(4)、C(4)-C(5)键则逐渐伸长,分别由反应物的1.397Å、1.390Å→过渡态TS1的1.423Å、1.421Å→中间体M1的1.478Å、1.474Å,可见C(3)-C(4)、C(4)-C(5)逐渐转变为单键。C(2)-C(3)、C(5)-C(6)键逐渐缩短,由反应物的1.397Å、1.397Å缩短至M1的1.358Å、1.352Å。邻苯二酚中的C(4)由sp2杂化变为sp3杂化,芳环的共轭性被破坏,H(7)逐渐偏离芳环分子平面,与芳环的夹角逐渐增大,由0.0°→12.296°→56.518°。
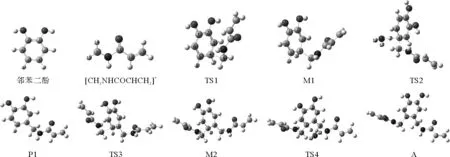
图3 反应物、中间体、过渡态和产物结构图Fig.3 Optimized structure of reactants, products and transition states
脱质子过程中,H2O分子(水是哪来的?乙醇不能帮助脱质子吗)以O原子端与中间体M1的H(7)逐渐接近,形成过渡态TS2,在水分子作用下H(7)逐渐脱离,生成一烷基化产物P1。过渡态TS2中,H(7)与芳环平面的夹角为92.25°几乎垂直于芳环,O(16)-H(7)距离为1.310Å,较水分子的O—H键(0.970Å)长;C(4)-H(7)键由中间体M1的1.103Å伸长至过渡态TS2的1.360Å,然后H3O+逐渐脱离至C(4)-H(7)键完全断裂,生成P1。H3O+为三角锥形,具有C3V对称性,O—H键长0.978Å,键角110.716°。由TS2至P1,C(4)-C(9)键缩短,由1.591Å变为1.536Å;芳环的C(3)-C(4)、C(4)-C(5)键也缩短,分别由1.437Å、1.426Å至1.401Å、1.393Å;C(2)-C(3)、C(5)-C(6)键则伸长,分别由1.375Å、1.379Å→1.390Å、1.397Å。可见产物P1中苯环中C—C键长的差异不大,恢复共轭性。
亲电取代反应中,亲电试剂优先进攻带负电荷较多的原子。单取代完成后,反应体系中同时存在邻苯二酚与一烷基化产物P1。由表1可知,P1中C(5)净电荷为-0.806,较C(3)(-0.067)、C(6)(0.050)占明显优势,也比邻苯二酚中C(4)或C(5)(-0.124)的电荷密度高很多,此时亲电试剂更易选择进攻单取代产物P1的C(5)位,形成二取代产物,而不是选择邻苯二酚中C(4)、C(5),生成单取代产物。二取代的C(5)-C(19)键形成过程与一取代类似。邻苯二酚被二取代后,C(3)(-0.084)与C(6)(-0.082)的净电荷低于邻苯二酚中C(4)、C(5)的静电荷,且其空间位阻较大,难以继续发生亲电取代。
2.5 反应势垒
图4(a)、4(b)分别为一取代、二取代反应过程势能变化,分别以两个过程的反应物为势能零点。一烷基化反应过程中R1经过渡态TS1至中间体M1,是吸热反应,活化能为61.07kJ/mol。中间体M1经过渡态TS2脱去质子,生成一烷基化物P1,活化能为39.69kJ/mol。一取代反应体系共放热35.41kJ/mol。二取代过程R2由过渡态TS3至中间体M2,活化能为44.31kJ/mol,中间体M2经过渡态TS4脱去质子,生成最终产物N-(3、4-二羟基-6-丙烯酰胺甲基苄基)丙烯酰胺,活化能为31.51kJ/mol。二取代过程体系共放热74.04kJ/mol。比较一取代、二取代反应过程势能变化可以看出,二取代活化能为44.31kJ/mol较一取代活化能61.07kJ/mol低16.76kJ/mol,且二取代释放的总能量更多,因此亲电试剂更易与P1反应,生成二取代产物。
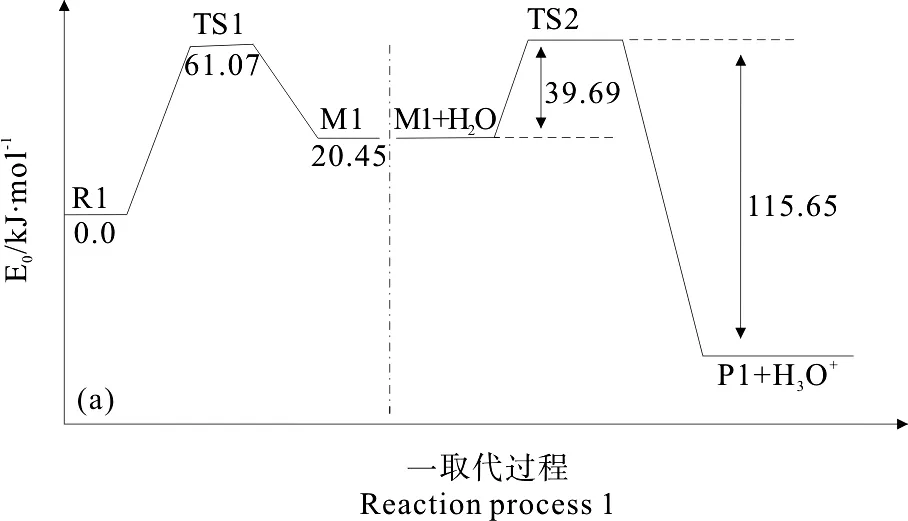
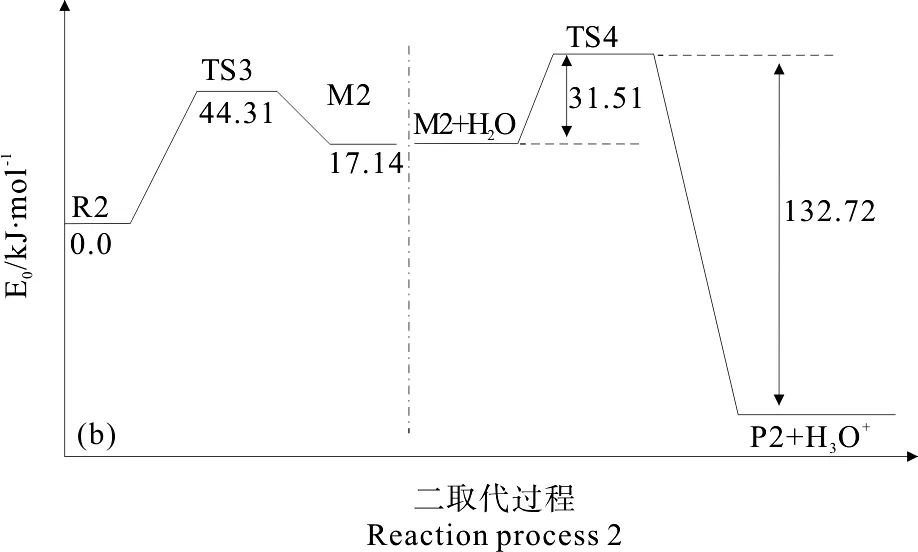
图4 邻苯二酚与[CH2NHCOCHCH2]+烷基化反应势能变化Fig.4 Potential energy profiles of alkylation of pyrocatechol with [CH2NHCOCHCH2]+ positive ion
综上,从热力学角度和静态原子电荷布居两方面的分析可知,二元苯酚和N-羟甲基丙烯酰胺的烷基化反应,以二烷基化为优势产物,与实验过程中只制得二取代产物一致。
3 结语
通过密度泛函计算,分析了邻苯二酚与N-羟甲基丙烯酰胺反应历程中过渡态、中间体的分子结构、活化能、前线轨道组成、反应途径及原子电荷等。从静态角度分析分子的电子结构表明,一取代产物芳环上碳原子C(5)为优势活性位点,且电荷密度高,有利于亲电试剂进攻,继续进行二取代反应;从热力学角度分析,二取代活化能低于一取代活化能,且二取代反应体系释放的总能量大于一取代反应的两倍,因此反应不是终止于一取代产物,更倾向于发生二取代反应。从动态和静态两方面均能够合理地解释了实验现象,并发现水分子在脱质子反应过程中起重要作用。
[1] 彭必先, 王俊莲, 彭争宏, 等. 辣椒素同系物合成、辣度及海洋生物防污性能研究 [J]. 中国科学: 化学, 2011, 41(10): 1646-1654.
[2] Xu Q W, Barrios C A, Cutright T, et al. Evaluation of toxicity of capsaicin and zosteric acid and their potential application as antifoulants [J]. Environ Toxicol, 2005, 20(5): 467-474.
[3] 史航, 王鲁民. 含辣椒素防污涂料在海洋网箱网衣中应用研究 [J]. 化工新型材料, 2004, 31(11):54-56.
[4] 杜伟娜. 高纯度人工合成辣椒素在防污涂料中应用效果的研究 [J]. 上海涂料, 2009, 47(8): 1-3.
[5] 李艳, 孔学, 王加宁, 等. 辣椒碱的合成及其防污性能研究 [J]. 安徽农业科学, 2010, 38(22): 11764-11765.
[6] 丛巍巍, 于良民, 姜晓辉, 等. 新型防污剂的合成、生物毒性及防污性能的研究 [J]. 材料导报B:研究篇, 2011, 25(2): 37-41.
[7] 闫雪峰, 于良民, 姜晓辉. 新型防污剂辣素衍生物的合成、抑菌性及防污性能研究 [J]. 中国海洋大学学报: 自然科学版, 2013, 43(1): 64-67.
[8] 何奕工, 舒兴田, 龙军. 正碳离子和相关的反应机理 [J]. 石油学报(石油加工), 2007, 23(4): 1-7.
[9] 聂小娃, 刘新, 宋春山, 等. H-ZSM-5分子筛上苯与乙醇和乙烯烷基化反应的理论研究 [J]. 催化学报, 2009, 30(5): 453-458.
[10] 郑天龙. 若干重要化学反应机理和溶剂效应的理论 [C]. 重庆: 西南大学, 2008.
[11] K Peter, C Vollhardt, Neil E Schore. 有机化学结构与功能 [M]. 戴立信, 席振峰, 王梅祥, 等译. 北京: 化学工业出版社, 2006: 5.
[12] 齐国鹏, 姜峰, 孙学文,等. Et3NHCl-AlCl3催化苯与1-十二烯烃烷基化反应机理的研究 [J]. 中国科学: 化学, 2010, 40(5): 461-466.
[13] 齐国鹏, 孙学文, 赵锁奇. [Bmim]Br-AlCl3催化苯与1-十二烯烃烷基化反应机理的研究 [J]. 有机化学, 2009, 29(12): 1963-1968.
[14] 孙学文, 赵锁奇. 超临界条件下离子液体催化苯与丙烯烷基化的反应机理研究 [J]. 化学学报, 2008, 66(4): 471-475.
[15] Sreekumar K, Sugunan S. Ferrospinels based on Co and Ni prepared via a low temperature route as efficient catalysts for the selective synthesis ofо-cresol and 2, 6-xylenol from phenol and methanol [J]. Journal of Molecular Catalysis A: Chemical, 2002, 185, 259-268.
[16] Sreekumar K, Sugunan S. A comparison on the catalytic activity of Zn1-xCoxFe2O4(x=0, 0.2, 0.5, 0.8 and 1.0)-type ferrospinels prepared via a low temperature route for the alkylation of aniline and phenol using methanol as the alkylating agent [J]. Applied Catalysis A: General, 2002, 230: 245-251.
[17] Krishna G B, Anup K T, Parashmani D, et al. Al-MCM-41 catalysed alkylation of phenol with methanol [J]. Journal of Molecular Catalysis A: Chemical, 2003, 197: 255-262.
[18] Namba S, Yashima T, Itaba Y, Hara N. Selective formation of p-cresol by alkylation of phenol with methanol over Y type Zeolite [M]. //Imelik B, Naccache C, Taarit Y B, eds. Catalysis by Zeolites. New York: [sin].1980: 105-111.
[19] 徐亚荣, 沈本贤, 徐新良, 等. FCC汽油噻吩类硫化物烷基化转移反应机理的量子化学 [J]. 石油学报(石油加工), 2011, 27(5): 806-811.
[20] Nie X W, Lin X, Song C S, et al. Bronsted acid-catalyzed tert-butylation of phenol, о-cresol and catechol: A comparative computational study [J]. Journal of Molecular Catalysis A: Chemical, 2010, 332: 145-151.
[21] Nie X W, Lin X, Gao L, et al. SO3H-functionalized ionic liquid catalyzed alkylation of catechol with tert-butyl alcohol [J]. Ind Eng Chem Res, 2010, 49, 8157-8163.
[22] Nie X W, Michael J J, Guo X W, et al. Reaction mechanism of tert-butylation of phenol with tert-butyl alcohol over H-β zeolite: An ONIOM study [J]. Catalysis Today, 2011, 165: 120-128.
[23] 付蓉, 卢天, 陈飞武. 亲电取代反应中活性位点预测方法的比较 [J]. 物理化学学报, 2014, 30(4): 628-639.
[24] Anslyn E V, Dougherty D A(著). 计国桢, 佟振合, 等译. 现代物理有机化学[M].北京: 高等教育出版社, 2009: 580.
责任编辑 徐 环
DFT Study of Pyrocatechol Alykation Reaction Mechanics
WU Xiao-Jing1,2, XIA Shu-Wei1,2, YU Liang-Min1,2, CHEN Guo-Bo1,2
(Ocean University of China, 1. The Key Laboratory of Marine Chemistry Theory and Technology, Ministry of Education; 2. College of Chemistry and Chemical Engineering, Qingdao 266100, China)
When capsaicin derivatives were synthesized from the alykation reaction of Binary phenol and N-methylolacryamide, as the ratio of Pyrocatechol to N-methylolacryamide changed, it was found that products were only dialkylation. The structures of reactants, transition states, intermediates and products were optimized by using DFT B3LYP/6-311G(d, p). The frequency, energies and charges were also analyzed. The calculated results show that the reaction includes four steps. The activation energies (Ea) of the four steps are 61.07 kJ/mol、39.69 kJ/mol、44.31 kJ/mol and 31.51 kJ/mol respectively. The whole reaction is exothermic except that the electrophilic attack step is endothermic. The reaction energy barrier of the dialkyl reaction is 44.31 kJ/mol, lower than 61.07 kJ/mol of monoalkyl reaction. In addition, the Mulliken charge datas show that the mulliken charge of the active atom increases after the pyrocatechol being substituted. As a result it is easier to be attacked by the positive ion.
pyrocatechol; reaction Mechanism; friedel-crafts alykation; density functional theory
国家自然科学基金项目(20677053)资助
2014-06-05;
2014-08-19
吴晓静(1989-),女,硕士生。E-mail:798696227@qq.com
❋❋ 通讯作者: E-mail: shuweixia@ouc.edu.cn
O643.12
A
1672-5174(2015)08-071-05
10.16441/j.cnki.hdxb.20140190
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
化学分析计量(2021年6期)2021-06-22 09:12:52
闽南师范大学学报(自然科学版)(2020年1期)2020-06-05 06:19:00
生物技术通报(2019年9期)2019-09-18 10:05:32
电脑知识与技术(2018年3期)2018-03-21 09:27:04
石油石化绿色低碳(2018年5期)2018-03-20 04:41:21
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
石油炼制与化工(2016年6期)2016-04-06 22:54:21
合成化学(2015年2期)2016-01-17 09:04:08
