
生物等效性研究中的统计学问题
2015-03-09 12:57沈卓之杨
中国卫生统计 2015年4期
沈卓之杨 珉
生物等效性研究中的统计学问题
沈卓之1杨 珉2
生物等效性(bioequivalence,BE)评价是药代动力学的重要课题之一,也是近年来统计学应用的热点领域[1-5]。尽管一般药物BE评价的统计方法已基本趋于成熟,但现有的统计方法尚有值得商榷之处,且仍有文献报道中存在试验设计和统计处理的缺陷。本文拟对生物等效性研究中存在的几个统计学问题进行文献复习讨论。
试验设计
生物等效性研究中的试验设计主要有交叉设计、平行设计、平衡的不完全区组设计等几种方法。只要实际操作上可行,各国指导原则均建议生物等效性试验尽量采用交叉设计的方法[5]。
1.交叉设计(cross-over design)
在大多数生物等效性检验中,一般均采用交叉设计的方法,即每个病人或研究对象轮流接受每一种处理方法,他们在两个或多个时刻接受不同的处理[6]。这是由于生物利用度测量值的个体存在差异,而个体间的变异系数往往远大于个体内变异系数。交叉设计可以将个体间的变异分离出来,提高检验的敏感性和准确性,有效规避个体间变异给试验带来的偏倚[7]。
交叉设计有不同的设计类型,主要表现在不同顺序(sequence)不同阶段(period)的不同处理(treatment)的安排上。在生物等效性试验最基本且最常用的是两处理、两阶段、两序列的交叉设计,也称2×2交叉设计[8]。随着等效性试验的发展,多阶段交叉试验设计应运而生,该设计在一次试验中能同时比较多种处理因素,如试验包括3种药物(2个试验药和1个参比药)时,则宜采用3处理3阶段的3×3拉丁方试验设计[9]。多阶段交叉试验设计是将不同的研究对象按照事先设计好的不同的试验次序,在不同的时期逐次实施多种处理,以检验试验药和参比药是否具有生物等效性的一种设计方法。
平均生物等效性(average bioequivalence,ABE)或群体生物等效性(population bioequivalence,PBE)研究时不一定必须采用重复交叉设计,但个体生物等效性(individual bioequivalence,IBE)研究由于个体内方差以及个体与药物的交互作用,必须采用重复交叉设计[10]。而对于某些高变异性药物(highly variable drug),根据具体情况,除采用增加例数的办法外,也可采用重复交叉设计,对同一受试者两次接受同一制剂时可能存在的个体内差异进行测定。Patterson等建议[11],在不增加试验人次的情况下,采用重复交叉设计或组别-队列设计(group-sequencial design)[12],增大等效性试验的检验效能。换言之,重复交叉设计可以在相同的检验效能下,减少受试者数目,这将极大减少因扩大受试者数目而带来的伦理学问题。当然,它也有一定缺点:每个受试者需接受多种处理,因此需要较多的时间,受试者的失访或退出会影响整个试验。
对两种药物进行比较,常见的重复交叉设计有以下几种[13]:
(1)2×4重复交叉设计:在四个用药阶段中,两种药物按两个或多个顺序进行试验。常用设计有:

若以上顺序均采用,则为4×4重复交叉设计。
(2)2×3重复交叉设计:在三个用药阶段中,两种药物按两个或多个顺序进行试验。常用设计有:

(3)Balaam重复交叉设计:如果采用两阶段的交叉设计,则需要多个顺序才能得到个体内变异和个体与药物的交互作用。

2001年FDA指南指出,对于IBE的评价,若采用2×4设计,推荐(TRTR,RTRT)顺序;而对于2×3设计,则推荐(TRT,RTR)顺序。
对于FDA指南中的推荐,学术界提出的问题主要有以下几点:
(1)在总例数一定的前提下,哪种设计的有效性更高[14-15]。
(2)若与不同用药顺序的其他交叉设计相比较,如(TRRT,RTTR)、(TTRR,RRTT)、(TRR,RTT)、(TTR,RRT)等,何者为优[16-18]。
(3)若重复交叉设计中存在缺失数据,如何评价其PBE和IBE[19,20]。
对于以上问题,目前从统计学角度进行的研究尚不多。吕静静、于浩、陈峰等学者[21-23]采用Monte Carlo模拟研究对生物等效性试验的检验效能和样本含量估计作了较系统的研究。结果表明,4×4交叉设计的效率最高,其次为2×4交叉设计、2×3交叉设计、2×2交叉设计,平行组的设计效率不足交叉设计的一半,而Balaam设计的效率最低。因此,实际工作中,如果增加两个阶段时间的试验成本不是很高,且受试者有较好的依从性,推荐使用2×4设计或4×4设计来进行生物等效性试验。
2.平行设计
平行设计即完全随机设计[24-25],每组受试者例数相同,每个个体随机接受一种药物或药物的一种剂型。平行设计常用于Ⅱ、Ⅲ期临床试验,在生物等效性研究中并不常用。因为通常而言,观察次数相等时,检测产品的差异时用交叉设计比平行设计更加有效。但在以下情况下,平行设计优于交叉设计:(1)个体间变异性比个体内变异性小;(2)因药物半衰期较长,洗脱期相应较长,研究时间延长,增加了个体失访的机会,造成数据脱落;(3)受试者需多次进行血液采样,不易实施。
顺序效应(Sequence Effect)
顺序效应常与残留效应(carry-over effect)以及处理与阶段(时期)的交互效应相混淆[26-28]。残留效应是指:如果A、B两种药物均对下一阶段无影响或者影响相同,则AB与BA两种顺序的效应相等或相近,此时该效应混在阶段效应中,即可以忽略它的影响;如果A影响B,而B不影响A;或者A对B的影响不等于B对A的影响,则AB和BA两种顺序的效应不相同,此时就不能忽略其影响,且表现在第二阶段的处理效果中[29]。
顺序效应因其在一定程度上反映了残留效应的大小,常常被认为就是指药物残留效应,实际上这只是最常见的一种。除此以外,还有心理效应(psychological carry-over effect)、第一阶段用药导致耐药性而产生的撤退效应(withdrawals effect),以及病人身体状况因用药而改变所导致的遗留效应(nonuniform carry-over effect)等等[30]。它们均可通过合理的设计来去除(双盲法可去除心理效应,适当的效应去除时间可去除大部分其他种类的顺序效应,安慰剂一般不会和有效药物产生相互作用)。
在目前交叉设计常用的统计方法中,一般未考虑试验顺序对试验结果可能产生的影响,但这在很多情形下是不能忽略的。李晓松、张文彤等学者[31-32]运用多水平统计模型,在模型水平2随机部分拟合顺序效应的随机系数。顺序不同,试验结果在患者间的变异不同,即在患者间变异(即水平2方差)中进一步分解出顺序的效应。但若将此方法运用于生物等效性评价中,由于在水平2随机部分引入了顺序效应,势必会影响个体间变异的估计值,导致等效标度(criterion)的估计出现偏差,该问题的解决方法尚需进一步研究。
综上,无论是多阶段交叉设计、平行设计或是提出新的统计模型,在改善顺序效应的同时也都有不足之处,目前似乎尚未找到两全之策。正如Senn[33-34]所建议,是否存在顺序效应的影响应当基于生物学机制、药理和临床专业知识,只要所接受的处理不是内源性物质,不同处理间隔的洗脱期足够长,建议对顺序效应采取简化处理,而非单纯针对其建模。关键是在试验的设计阶段就应考虑到顺序效应的存在,减少统计分析时所设置的不合理的假设。
样本含量
ABE评价的仅仅是总体均数的差异,样本含量可直接通过公式估计这里Δ代表生物等效性的容许界值,例如log(1.25)或log(0.80);θ是log(μT/μR)或对数转换后两药效应的差别。当μT/μR大于1时,Δ取log(1.25),否则Δ取log(0.80)[37]。可见,与临床试验的样本含量估计一样,生物等效性试验的样本含量N也由以下三个基本因素决定:(1)显著性水平:通常取0.05;(2)检验效能,一般为80%;(3)变异性和差别(θ):两药ABE评价中检测指标的变异性越大,所需例数越多;且在不超出容许界值的假设下,θ值越大,所需例数越多[38]。

表1 ABE研究中不同变异度的样本含量标
PBE和IBE的评价统计量是均数和变异的综合指标,所以它们的样本含量只能通过模拟给出[39]。最理想的设计,是采用最少的受试者例数达到所需检验效能来判断两种制剂是否生物等效[40]。以下样本量推荐均来自2001年FDA正式发布的《生物等效性统计分析指导原则》[13]中采用Monte Carlo模拟研究估算出的样本含量。
1.ABE研究的样本含量
在不同国家颁布的生物等效性(ABE)研究指南中,对受试者例数的要求有所差别,美国为12~36例,欧盟须>12例,日本为20~30例,我国则要求18~24例健康男性,目前尚缺乏国际的一致性标准。但我国现行《新药审批办法》《药品注册管理办法》[41-42]均指出,对于高变异药物(个体内变异>30%),应当适当增加样本量,以保证试验有适当的检验效能,以避免出现不等效的假阴性结果。
2.PBE、IBE研究的样本含量
评价PBE、IBE的统计量是由效应值的均数、方差、个体内的方差以及个体与药物的交互作用组成,因此样本含量的估计是基于模拟数据产生的。该模拟数据的T与R的平均等效性在方差和个体-药物之间交互作用相同的情况下,其变异度不超过5%,样本的检验效能应达到80%~90%。
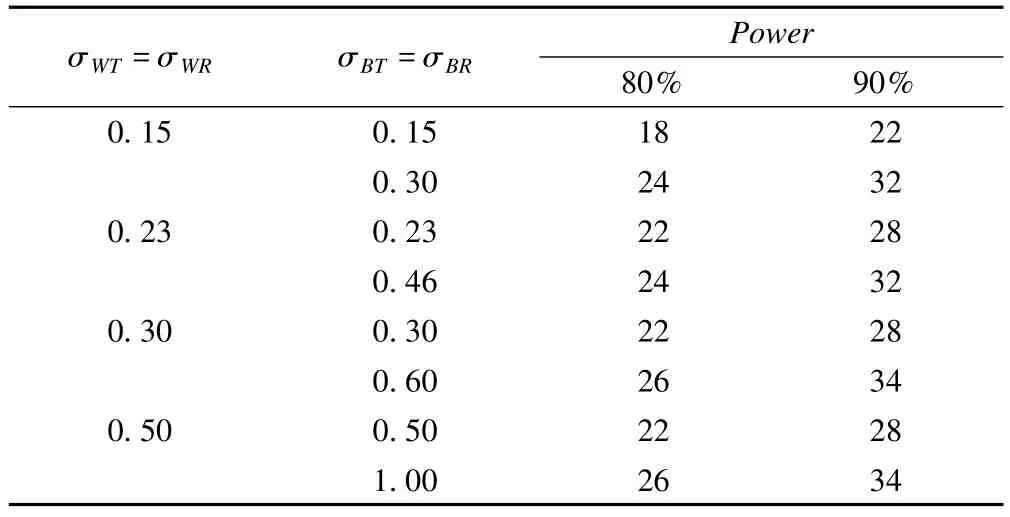
表2 PBE研究中不同变异度的样本含量
而IBE的样本含量因不同的个体内变异和个体与药物间的交互作用、设计而不同。需值得注意的是:FDA规定每一个序列的最小样本含量为6,所以当模拟结果的样本含量小于6时,在IBE评价试验中,应以6为每顺序的样本含量进行试验[43]。
对数变换
评价生物等效性的统计方法,其基本假设是数据服从或近似服从正态分布,这也是方差分析的前提条件[44-45]。当数据不服从正态分布时,常需进行对数转换来校正其对称性。这是因为生物等效性评价是以药代动力学为基础的,所依据的药物血液中时间-浓度公式为c(t)=Ae-λt[37]。当时间t=0时的药物浓度为A,λ是消减常率。只有当两药的时间-浓度函数相等,即cT(t)=cR(t)时,两药才能相互替换。这时的等效性可表为log(AT)-λT=log(AR)-λR。在等效假设下,总有期望值λT=λR,因而可用对log(AT)=log(AR)的假设检验获得等效性的推断。由此可知:(1)药代动力学参数以原始尺度进行分析,其效应不是可加性的,而对数转换可用加法模型进行处理;(2)AUC和Cmax通常为正偏态分布,且方差不齐,对其作对数变换可以改善其分布的偏性,缩小方差间的差异;(3)进行对数变换后,用方差分析和双单侧t检验,分析药代动力学参数的可靠性和检验效能更好。因此FDA指导原则中明确要求,生物等效性的统计分析应在药代动力学参数的对数尺度下进行。而生物等效性的评价是在还原原始尺度后的两药效比值及其90%可行区间下进行的。
然而有学者认为[24-45],FDA指南要求对所有参数均进行对数转换,这并非总是正确的。Patel[46]同样认为,将原始数据进行对数转换后再进行线性分析的策略并不是“科学”的方法。因为在BE研究中样本容量是有限的,即使采用对数变换也不能从根本上对一组数据是否符合正态分布作出可靠结论。
此外,根据以上等效评价原理,FDA要求,统计分析的结果要用对数尺度的,还需提供原始尺度的,即将对数结果取反对数转换。然而,原始尺度的对数转换有可能因为非线性函数的逆转换而引入偏差。例如,均值之比的点估计,若以对数尺度计算的结果取反对数来作为其原始尺度的结果,将会造成偏倚,并且总是正偏倚(positive bias)的。再有,将对数尺度上的标准差取反对数,显然并非其原始尺度上的标准差。结果应如何在原始尺度上进行表达,尤其是变异(方差)的表达,还需更进一步的研究。
值得注意的几个问题
1.关于普通t检验和双单侧t检验(two-one-sided test,TOST)
目前,我国ABE评价试验结果的统计分析,主要是用药动学参数AUC和Cmax经对数转换后以多因素方差分析进行显著性检验,然后对其几何均值进行双单侧t检验,以及计算90%置信区间的方法来评价和判断药物间的生物等效性[47]。方差分析法提供了其他评价方法都需用到的标准差,是包括双单侧t检验在内的其他评价方法的基础。对于方差分析和双单侧t检验的异同,已有文献报道[24]。在实际应用中,仅采用成组/配对t检验的情况仍时有发生,差异性t检验和双单侧t检验的意义和相互关系模糊不清,在此进行讨论。
成组/配对t检验是差异性检验,而双单侧t检验是等效性检验,这是本质完全不同的两种检验。成组/配对t检验的无效假设是两药有无差异,只在P<0.05时才认为两药差异有统计意义,但不一定不等效;P>0.05时认为两药差异无统计意义,但并不能认为两者等效。
而等效性检验意在确定两药差别无临床意义,其无效和备择假设是:
H0:(μA≠μB);H1:(μA=μB)。给定一个临床意义下的容许差值d后,得到以下式:
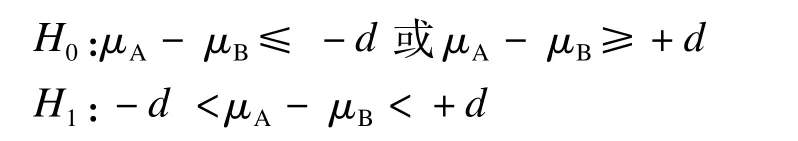
等效检验是对H0中的两个假设进行,故称双单侧t检验(TOST)。设定的无效假设是两药不等效,受试药在参比药一定范围之外,在P<0.05时说明受试药没有超过规定的参比药的高限和低限,可认为两药等效[37]。等效性检验离不开等效标准,差异显著性检验则与等效标准无关。要证明两药的药效相近,绝不能仅以P>0.05为依据,必须作双单侧t检验。
2.关于Tmax的分析
生物等效性评价的三个指标中,AUC、Cmax通常服从对数正态分布,相应的统计检验分析方法发展得比较成熟、完全。但作为反映药物吸收速率的指标Tmax,其重要性常有意或无意地被研究者忽略,统计检验分析方法也常照搬AUC、Cmax的方法。有学者认为,Tmax根据实测值得到,是一种离散的计数资料,服从参数Poisson分布[48],并不符合方差分析的假设基础,因而建立在方差分析上的双单侧t检验法和90%置信区间法不适用于Tmax的统计检验[24,49]。还有学者认为Tmax的分布性质是未知的,有时其数据太偏离正态分布,以致即使在处理平均值时也不能接受正态假设[50]。因此一般采用非参数方法(亦称为无分布统计)对Tmax进行检验,它可以对显著性水平做一个快速的近似估计。
尽管新药研究指导原则对于Tmax的统计分析作出了非参数检验的要求[51],但目前发表的生物等效性研究文章以及新药申报资料中仍有一部分按照双单侧t检验法和90%置信区间法对Tmax进行统计分析[52-53]。而在统计分析中,等效区间界值的取值范围,也常借用AUC、Cmax的取值范围(80%~125%或70%~143%),或自定取值范围,或没有说明等效界值范围,缺乏有力的证据支持。其实质是没有考虑Tmax的分布特点,机械重复AUC、Cmax的统计检验方法。
虽然采用非参数检验法的秩和检验考虑到了Tmax的分布,但由于秩和检验法是一种差异性检验而非等效性检验。当秩和检验法能作出两药Tmax存在差异的统计判断时,尚不能得到两药以Tmax为指标生物等效的统计结论[54]。考虑到后一情况在生物等效研究中更常见,而目前对于以Tmax为药代动力学指标来评价BE尚缺乏深入研究,因此有必要针对Tmax发展适当的统计检验方法,并探索相应的等效界值[55]。
另外,即使用非参数方法检验出两药间Tmax存在显著差异的结论,也可从以下两个方面考虑:(a)非参数方法的检验效能较t检验或F检验可能稍低;(b)一般的等效性检验主要还是针对AUC和Cmax的检验,对于Tmax,仅仅在药物释放快慢与临床疗效和安全性密切相关时才需要着重关注,故应当运用医学专业知识判断其在等效性检验中的权重[24,56]。
[1]Chow SC,Liu J.Design and analysis of bioavailability and bioequivalence studies.Vol.27.2009:Chapman&Hall/CRC.
[2]Popovi,J,M ikov M,Sabo A.Evaluation of statistical power function for various diclofenac bioequivalence trials with different subject numbers.European Journal of Drug Metabolism and Pharmacokinetics,2009,34(2):85-91.
[3]Arieta AG.Bioequivalence assessment of inhalation products:Interchangeability,study design and statisticalmethods.Pulmonary pharmacology&therapeutics,2010,23(3):156.
[4]O′Connor D,AdamsWP,Chen ML.Role of pharmacokinetics in establishing bioequivalence for orally inhaled drug products:workshop summary report.Journal of aerosolmedicine and pulmonary drug delivery,2011,24(3):119-135.
[5]Pathak R,Swamy SGDV.A Bief Review-study Guidelines for Conducting Bioavailability I/Bioequivalence(BA/BE)Studies and its Statistical Analysis.Inventi Impact:Pharmacokinetics&Pharmacodynam ics,2012.
[6]Ratkowsky DA,Evans MA,Alldredge JR.Cross-over experiments:design,analysis,and application.Vol.135,1993:CRC.
[7]姚晨,陈峰.交叉试验设计资料的等效性检验.中国临床药理学杂志,2001,17(4):294-297.
[8]Vuorinen J.A practical approach for the assessmentof bioequivalence under selected higher-order cross-over designs.Statistics inmedicine,1997,16(19):2229-2243.
[9]Chow SC,Liu JP.On assessment of bioequivalence under a higherorder crossover design.Journal of Biopharmaceutical Statistics,1992,2(2):239-256.
[10]Hyslop T,Iglew icz B.Alternative cross-over designs for individual bioequivalence,2001.
[11]Patterson SD,Zariffa NMD,Montague TH.Non-traditional study designs to demonstrate average bioequivalence for highly variable drugproducts.European Journal of Clinical Pharmacology,2001,57(9):663-670.
[12]Jennison C,Turnbull BW.Group sequential methods with applications to clinical trials.2000:CRC Press.
[13]FDA(2001)Guidance for Industry:Statisticalapproaches to establishing bioequivalence.Rockville,MD:US Food and Drug Adm inistration Center for Drug Evaluation and Research.
[14]Stefanescu C,Mehrotra DV.A More Powerful Average Bioequivalence Analysis for the 2?×?2 Crossover.Communications in Statistics-Simulation and Computation,2007,37(1):212-221.
[15]Rani S,Pargal A.Bioequivalence:An overview of statistical concepts,2004.
[16]Esinhart,JD,Chinchilli VM.Extension to the use of tolerance intervals for the assessment of individual bioequivalence.Journal of Biopharmaceutical Statistics,1994,4(1):39.
[17]Haidar,S,Davit B,Chen ML.Bioequivalence Approaches for Highly Variable Drugs and Drug Products.Pharmaceutical research,2008,25(1):237-241.
[18]Chow SC,Shao J,Wang H.Individualbioequivalence testing under2 ×3 designs.Statistics in medicine,2002,21(5):629-648.
[19]Yueyuan M,X Yongyong,G.Xiue.Department of Health,Statistics,Fourth M ilitary Medical University(710032),Xian;Bioequivalence Assessment with M issing Data Using MCMC.Chinese Journal of Health Statistics,2004,4.
[20]Donner,A.,W.W.Hauck,G.Zou.The impact ofm issing values in the concentration-time curve on the assessment of bioequivalence.Pharmaceutical Statistics,2005,4(2):91-99.
[21]于浩,吕静静,陈峰.平均生物等效性试验设计方法评价.中国卫生统计,2004,21(6):332-334.
[22]陈峰,于浩,吕静静.生物等效性评价的统计分析方法.中国临床药理学与治疗学,2004,9(8).
[23]吕静静,陈峰,崔永春.个体生物等效性检验的样本含量估计.中国临床药理学与治疗学,2007,12(2):204-207.
[24]杨进波,赵明,赵德恒.生物等效性试验中的统计学相关问题讨论.2005,国家食品药品监督管理局药品审评中心
[25]杨进波.关于平行设计生物等效性试验几个问题的探讨.中国临床药理学杂志,2008(6):479-480.
[26]Zintzaras,E.The existence of sequence effect in cross-over Bioequivalence trials.European Journal of Drug Metabolism and Pharmacokinetics,2000,25(3):241-244.
[27]D′Angelo,G.,D.Potvin,J.Turgeon.Carry-over effects in bioequivalence studies.Journal of Biopharmaceutical Statistics,2001,11(1-2):35-43.
[28]Liu,H.,J.P.Wang,H.Chen.Assessment of Individual bioequivalence in s-sequence four-period crossover design.Asian Journal of Drug Metabolism and Pharmacokinetics,2003,2004,4(1):43-48.
[29]Senn,S.,G.D′Angelo,D.Potvin.Carry-over in cross-over trials in bioequivalence:theoretical concerns and empirical evidence.Pharmaceutical Statistics,2004,3(2):133-142.
[30]Woods,J.R.,J.G.W illiams,M.Tavel.The two-period crossover design in medical research.Annals of internal medicine,1989,110(7):560.
[31]李晓松,张文彤.多水平模型在交叉设计资料分析中的应用.中国卫生统计,1999,16(5):273-274.
[32]张文彤,李晓松.交叉设计中顺序效应的影响及其分析方法.数理医药学杂志,1999,12(4):305-306.
[33]Senn,S.Controversies concerning random ization and additivity in clinical trials.Statistics in medicine,2004,23(24):3729-3753.
[34]Senn,S.Cross-over trials in Statistics in Medicine:the first‘25’years.Statistics in medicine,2006,25(20):3430-3442.
[35]Ganju,J.,A.Izu,A.Anemona.Sample size for equivalence trials:A case study from a vaccine lot consistency trial.Statistics inmedicine,2008,27(19):3743-3754.
[36]于莉莉,夏结来,蒋红卫.三种不同方法估算等效性检验的样本量与检验效能.中国卫生统计,2006,23(4):336-338.
[37]Julious,S.A.,Sample sizes for clinical trials.2009:CRC Press.
[28]Endrenyi,L.,L.Tothfalusi.Sample Sizes for Designing Bioequivalence Studies for Highly Variable Drugs.Journalof Pharmacy&Pharmaceutical Sciences,2011,1(15):73-84.
[39]Chen,K.W.,S.C.Chow,G.Li.A note on sample size determination for bioequivalence studies with higher-order crossover designs.Journal of Pharmacokinetics and Pharmacodynamics,1997,25(6):753-765.
[40]Chow,S.-C.,J.Shao,H.Wang.Sample Size Calculations in Clinical Research.Statistical Papers,2011,52(1):243-244.
[41]国家药品监督管理局,新药审批办法,1999.
[42]国家药品监督管理局.药品注册管理办法(试行).中国新药杂志,2002,12:1003-3734.
[43]Chow,S.-C.,J.-P.Liu.On assessment of bioequivalence under a higher-order crossover design.Journal of Biopharmaceutical Statistics,1992,2(2):239-256.
[44]Liu,J.P.,C.S.Weng.Estimation of direct formulation effect under log-normal distribution in bioavailability/bioequivalence studies.Statistics in medicine,1992,11(7):881-896.
[45]Lee,Y.J.,Y.Kim,M.Lee.Analysis of bioequivalence study using a log-transformed model.JOURNAL-PHARMACEUTICAL SOCIETY OF KOREA,2000.44(4):308-314.
[46]Patel,H.Dose-response in pharmacokinetics.Communications in Statistics-Theory and Methods,1994,23(2):451-465.
[47]韩伟,姜晶梅.生物等效性统计评价方法概述.中国卫生统计,2010(4):441-445.
[48]Basson,R.P.,A.Ghosh,B.J.Cerimele.Why rate of absorption inferences in single dose bioequivalence studies are often inappropriate.Pharmaceutical research,1998,15(2):276-279.
[49]刘晓东,杨劲.生物等效性评价的若干问题的探讨.中国临床药理学与治疗学,2000,5(3):248-252.
[50]Park,S.,E.Clarkson,M.A.Kupinski.Efficiency of human and model observers for signal-detection tasks in non-Gaussian distributed lumpy backgrounds,2005.
[51]国家食品药品监督管理局.化学药物制剂人体生物利用度和生物等效性研究技术指导原则.2005,国家食品药品监督管理局.
[52]张琰,杨蕾,刘梅.苯磺酸氨氯地平片在健康人体的药动学及生物等效性评价.第四军医大学学报,2006,27(2):172-175.
[53]孙伟,马霖,刘雅丽.多糖铁胶囊的人体生物等效性研究.中国药房,2004,15(3):167-168.
[54]Wijnand,H.P.Bioequivalence revisited:non-parametric analysis of two-period cross-over studies.Computer Methods and Programs in Biomedicine,1993,40(4):249-259.
[55]Chen,M.L.,L.Lesko,R.L.W illiams.Measures of exposure versus measures of rate and extent of absorption.Clinical pharmacokinetics,2001,40(8):565-572.
[56]Basson,R.P.,B.J.Cerimele,K.A.DeSante.Tmax:an unconfounded metric for rate of absorption in single dose bioequivalence studies.Pharmaceutical research,1996,13(2):324-328.
(责任编辑:邓 妍)
1.重庆市疾病预防控制中心慢病所(400042)
2.四川大学华西公共卫生学院卫生政策和管理系
猜你喜欢
今日农业(2021年12期)2021-11-28
新世纪智能(数学备考)(2021年9期)2021-11-24
中学生数理化·高一版(2021年2期)2021-03-19
新世纪智能(数学备考)(2020年9期)2021-01-04
语数外学习·高中版上旬(2020年8期)2020-09-10
初中生世界·八年级(2019年6期)2019-08-13
中学生数理化·高一版(2018年10期)2018-11-08
领导决策信息(2018年16期)2018-09-27
数学学习与研究(2017年3期)2017-03-09
小学生导刊(低年级)(2016年9期)2016-10-13
