
高通量计算在锂电池材料筛选中的应用
2015-02-25 02:11凌仕刚肖睿娟欧阳楚英陈立泉
中国材料进展 2015年4期
凌仕刚,高 健,褚 赓,黄 杰,肖睿娟,欧阳楚英,李 泓,陈立泉
(1.中国科学院物理研究所,北京 100190)(2.江西师范大学,江西 南昌 330027)
特约专栏

第一作者:凌仕刚,男,1988年生,博士研究生
高通量计算在锂电池材料筛选中的应用
凌仕刚1,高健1,褚赓1,黄杰1,肖睿娟1,欧阳楚英2,李泓1,陈立泉1
(1.中国科学院物理研究所,北京 100190)(2.江西师范大学,江西 南昌 330027)
摘要:先进电池技术是未来十年世界各国前沿技术竞争的制高点,锂电池由于具有电压高、比能量高、充放电寿命长、工作温度范围宽等优点,已成为许多移动电子产品、电动汽车以及风电和光伏电储能的首选。因此,更快的开发出能量密度高、安全性好、充放电速度快的高性能锂电池材料十分必要。借助高通量计算方法,通过设置合理的筛选条件,可加快锂电池材料的研发。通过使用自主编写的计算软件及高通量自动化计算流程,以无机材料晶体结构数据库中现有的结构为基础,针对电极材料和固体电解质材料分别应具有的特点,实现了对锂电池材料的初步筛选。计算结果与现有的实验数据汇集到一起,形成了庞大的数据库。进一步借助统计方法进行数据挖掘,有望揭示材料的结构-性能关系及内在物理规律,缩短材料从研发到应用的全过程,为开发全新电池材料乃至电池体系提供有力支持。
关键词:锂电池材料;高通量计算;热力学计算;键价和方法
1前言
能源的清洁高效利用离不开高性能储能器件。从铅酸电池、镉镍电池到镍氢电池,再到锂离子电池,化学电源技术得到了迅猛发展,能量密度也大幅提高。与其他蓄电池比较,锂电池具有电压高、比能量高、充放电寿命长、无记忆效应、无污染、快速充电、自放电率低、工作温度范围宽和安全可靠等优点[1]。锂电池的平均比能量是镉镍电池的2.6倍、氢镍电池的1.75倍,其工作电压为3.6 V,是镉镍、氢镍电池的3倍[2]。在民用领域,锂电池已经被广泛应用于各种电子产品、电动工具[2],并已成功应用于电动车、混合动力车等[3],同时锂电池或钠电池体系也是风电和光伏电储能的首选方案[4]。
当前,清洁高效的能源储存技术已成为工业可持续发展的核心关键技术之一,因此研发具有更高能量密度、更高功率密度、更长寿命、更加安全的可充放锂电池储能器件十分必要。现有锂电池正极材料体系主要包括层状材料(如LiCoO2)、尖晶石结构材料(如LiMn2O4)以及橄榄石结构材料(如LiFePO4)[5]。这3种材料的嵌锂电位平台分别为3.9 V、4.1 V和3.6 V,其容量分别为130~140 mAh·g-1、120~130 mAh·g-1和150~160 mAh·g-1。在这些材料体系基础上,还有其他通过改性的正极材料:比如层状的Li(NixCoyMn1-x-y)O2材料,其比容量能达到200 mAh·g-1甚至更高[6];通过对LiMn2O4材料掺杂,可以提高其电位到4.5 V[7]。然而,由于这些材料体系的结构稳定性、可转移电子和离子数的限制,经过近20年的研究,其能量密度已经接近其理论极限。要进一步提高电池的能量密度等性能,必须探索出全新的锂电池正极材料体系。
就负极材料而言,目前的炭负极材料的应用潜力也已经接近其理论极限,且应用过程中面临着各种技术挑战与难题。比如,石墨负极材料表面在使用过程中SEI(Solid Electrolyte Interphase)膜生长的控制、首次充放电效率的提高、对正极提供的Li源的消耗导致可逆容量损失等[8]。这些难题是炭负极材料本征特性带来的,要彻底解决这些问题,必须使用全新的负极材料体系。硅负极材料虽然理论容量高达4 200 mAh·g-1,但其嵌Li过程的体积膨胀(~400%)导致其循环性能的控制非常难,循环过程中Si和Li的反复合金化/去合金化过程的机械分裂和粉末化,将导致材料结构的坍塌,进而使得材料的容量急剧下降[9]。而Si材料嵌Li过程的体积膨胀也是材料的本征属性,难以从根源上彻底解决,因此迫切需要寻找性能更加优越的负极材料。
除了提高电池的能量密度,锂电池的安全性能也急待提高。电池的安全性能在很大程度上和使用的液态有机电解液体系有关。使用固体电解质体系的全固态锂离子电池,是从根源上解决电池的安全性问题的方案[10-11]。使用固体电解质材料体系的一个最大问题是锂离子在固体电解质材料中的离子输运很慢。一般来说,锂离子电导率在固体电解质中要比在液体电解液中低1~2个数量级[12]。最近,日本一研究小组在NatureMaterials[13]上报道了他们开发的Li10GeP2S12材料, 该材料在室温下锂离子的电导率可以达到10-2S/cm,说明固体电解质材料在锂离子电导率方面已能满足电池应用的要求,但该材料在自身的稳定性、和电极材料界面的兼容性等其它方面还存在一些问题[14],开发兼具高离子电导率、高稳定性、高机械强度的固体电解质材料势在必行。
由此可见,当前锂电池产业面临的瓶颈问题是现有的电池材料体系的本征特性,决定了现有电池体系的潜力有限,不能满足高能量密度、高功率密度、高安全性能、低成本等多方面的需求。而要从根源上解决这些问题,必须开发出全新的锂电池材料,进而开发出全新的锂电池体系,如全固态锂电池体系[11],Li-O2电池体系[15],Li-S电池体系[16],Li-CO2电池体系[17]等。然而,传统的材料研发方法周期很长,过去20年对锂电池材料的开发沿用“炒菜”式的实验组合方法,仅开发了6种能够实际应用的电池材料,其它工作大多是对这几种材料的改性或修饰。近年来,随着研究人员对锂电池中热力学及动力学过程理解的不断深入,加之计算机运算能力的飞速发展,理论计算已被有效的应用到电池材料的研究中[18-20]。采用理论模拟的方法一方面可以对已有的材料进行模拟和计算,分析它们的微观物理机理,对相应的物理现象加以解释,为实验提供理论根据;另一方面能预测未知材料的性能,为实验指导方向。对锂电池材料的计算具体包括:计算材料的电子结构[21],分析其电子导电性[18];模拟离子在材料中扩散,分析材料的离子导电性[22];通过模拟充、放电过程,计算脱/嵌锂电位,分析材料的体积和结构变化,以此来说明它的结构稳定性和循环性[23];研究电极材料表面以及表面和电解液之间相互作用,探索电池材料失效的机理等[24]。自然界中有着成千上万的化合物,已经研究过的只是少数。对已存在物质的基本性质进行计算,有可能从中筛选出可以用于锂电池的新材料。对每种物质的计算无论是从方法选择、计算参数、数据输入等方面都具有一定的相似性,因此采用自动化的操作来调用软件、输入数据、控制运算步骤、分析计算结果就十分必要。这种高度自动化的运算过程称为高通量计算。高通量计算具有高效能、可并行、可扩展等优点,有助于我们设计一些筛选材料体系的方案。借助高通量的计算,可以有目标的去发现一些新材料体系,同时研究这些新体系,进一步增加对锂电池材料的认识,从而形成不断深入的研究和开发过程,最终有助于我们更快的开发出能量密度高、安全性好、充放电速度快的高性能锂电池材料。
2高通量计算发展概述
高通量方法在生物学、药学和化学领域已得到广泛的应用,例如高通量测序(High-Throughput Sequencing)方法可以并行的合成多达50万条DNA分子并进行序列测定[25-26];高通量筛选(High-Throughput Screening)技术则通过分子水平和细胞水平的实验,以自动化操作同时制备、检测成千上万的样品,实现药物分子的筛选[27]。在材料科学中,1995年Xiang等人[28]将薄膜沉积和物理掩膜技术相结合,在200 μm×200 μm大小的样品上制备出一系列不同组分的BiSrCaCuO和YBaCuO超导薄膜,实现了对材料的组合制备和高通量筛选,为新材料的发现提供了一种新的思路和方法。

在锂电池材料设计开发的过程中,采用高通量计算的理论方法始于MIT的Ceder研究组,他们从2010年开始开展了称之为“锂离子电池材料基因工程”的研究[33]。该方法通过对含聚阴离子XO4(X=P,S,As,Si)的化合物中元素的替代,来产生新化合物,计算了其能量密度、电压、脱Li后的体积变化等参数,据此筛选新材料[34]。他们从自然界存在的矿物Sidorenkite结构出发,对其进行元素替换,构造出270多种组分和结构,并计算其性能,从中筛选出了几种材料:Li3Mn(CO3)(PO4)、Li2V(CO3)(PO4)、Li3V(CO3)(SiO4)等。应用类似办法,他们还对含有(PmOn)阴离子团的各种过渡金属与Li的化合物进行了筛选[35]。同时,在上述研究过程中,他们也初步发展了一套系统的性能计算、数据生成、数据分析的办法[33]。日本的Tanaka研究组将第一性原理分子动力学模拟与机器学习技术相结合,研究了LISICON类型材料的离子输运,通过将理论计算结果与实验数据库结合,可有效预测LiO1/2-AOm/2-BOn/2体系不同组分的离子电导率,有助于加快固态电解质的开发[36]。
上述研究工作中所用到的高通量计算方法都具有图1[33]所示的共同特点。首先从外部的结构数据库中选择数据,产生可被计算软件调用的输入文件,并进行计算得到材料相应的性质数据,运算结果保存到数据库以备进一步的分析,获得的新知识可扩充原先的数据库并有助于更为准确的数据选择。由此可见,形成并完善一套程序化的高通量计算流程,把各种计算软件包、编写的单一功能的计算程序或指令(如输入文件生成、材料性质模拟、结果分析程序等)和计算硬件设备等关联起来,使得整个过程能够自动完成,是实现高通量计算提高整体效率的关键。上面提到的由Curtarolo等人设计的AFLOW[30]和由Ceder小组设计的计算流程[33]均实现了晶体结构数据库与第一性原理计算程序VASP[37]的自动调用功能,从而通过高通量计算获得材料的形成能等热力学数据以及电子结构的信息。但由于筛选不同类型的材料时所关注的性质往往是不同的,希望调用的材料数据库、模拟方法、分析方法都可能有所变化,所以针对各种不同的材料还需设计相应软件以实现特定的自动化运算流程。

图1 高通量计算筛选材料的数据流程图[33]Fig.1 Data flow diagram of high-throughput computational screening[33]
目前对于锂电池材料的高通量筛选,国内外的研究工作都处于起步阶段,尚存在以下问题:①偏重于材料热力学性质,如形成能、热稳定性等的计算,缺乏对锂离子输运这一重要的动力学性能的考虑;②筛选所用的约束条件缺乏针对性和层次性,造成大量重复及不必要的计算;③计算采用的多是基于密度泛函的第一性原理方法,尚未结合其他更多的计算方法或软件;④对计算得到的大量数据尚未进行有效的挖掘。事实上,材料的性能完全由材料的结构和组分两个因素决定,通过大规模的计算,挖掘出材料结构、组分和材料性能之间的联系,构建出材料的“构效关系”,也是高通量计算的重要目标之一。通过对筛选过程设立有针对性的外部约束条件,包括应用要求约束、基本物理规律约束、温度压强等外部条件,构建材料组分和结构模型。通过高通量计算技术来计算这些结构模型对应的化合物的各种性能,并反馈到结构性能数据库,用于研究锂电池材料中的物理问题,同时筛选出满足应用约束条件的材料。并通过各种实验方法制备这些材料,然后测试和表征这些材料的物理化学性能,最终达到理解锂电池材料物理问题,并发现新材料的目的。
可以预见在未来几年内,应用高通量计算方法,结合实验研究,将大大加速我们对锂电池材料的研发,加深对锂电池中各种问题的理解,揭示材料的结构-性能关系及内在物理规律,从而缩短材料从研发到应用的全过程。
3锂电池材料性能的计算方法
在高通量计算中,可以根据所筛选材料的特点综合使用计算材料学中的各种技术方法和手段,包括热力学计算、第一性原理计算、分子动力学模拟、蒙特卡罗模拟等。对于锂电池而言,电极材料的储锂容量、嵌锂电位、电子导电性、锂离子扩散率、脱嵌锂过程中的体积变化、结构相变,电解质材料的离子电导率、电化学窗口宽度、结构稳定性,电解质与电极之间界面的化学稳定性等因素都会影响到电池的性能。上述的材料性质都有相应的计算方法,根据各种方法的原理,会得到不同计算精度的结果,各种方法适当组合,可以构建出合理的材料筛选流程。
3.1理论储锂容量
电极材料的储锂容量与可转移电子数、可转移离子数有关。对于给定的电极材料,其理论储锂容量可由式(1)计算得到[38]:
Capacity=nF/3.6M(mAh·g-1)
(1)
其中,n为每摩尔所能转移的电子数目,M为摩尔质量(g·mol-1),F为法拉第常数。因此,根据电极反应中可以参与反应的Li+及电子的数目,即可得到电极材料的理论储锂容量,可用作电极材料初步筛选的参数之一。
3.2嵌锂电位
电极材料在脱嵌锂过程中的平均电位与电极反应的吉布斯自由能ΔrGΘ有关,如式(2)所示:
ΔrGΘ=-nEF
(2)
其中,E即为电极反应的热力学平衡电压。一方面,吉布斯自由能的数据可以从热力学实验数据手册[39]中获得,对于缺少实验数据的反应,可以通过第一性原理计算得到[40]。
3.3离子输运
锂电池中核心的动力学过程是锂离子的输运,离子传输的快慢和路径直接影响电池充放电的性能。在固体和液体中,离子的传输是通过离子向近邻位置随机跳跃的扩散过程实现的。离子在运动过程中会受到由周围离子形成的势场的阻碍,这种阻碍的大小可以用离子扩散活化能表示。对材料中离子输运的计算方法可分为基于几何结构[41-43]、基于成键配位[44]和基于能量变化[19,22]3类。基于几何结构的方法是分析晶格中存在的可供Li+运动的几何空间,能定性理解Li+在晶格中运动的难易程度。基于成键配位的键价理论[44]考虑了与Li+成键的阴离子对Li+运动的制约,通过引入基于Morse势的势能函数,并更进一步考虑Li+与其他阳离子的库仑排斥势,可以得出Li+在晶格中形成连通路径时所需要越过的势垒大小[45],由于该方法中使用的Morse势为经验势,因此势垒的计算属于半定量性质。更为精确的基于能量的计算,可借助过渡态理论,通过DFT计算来得到,例如采用爬坡弹性能带(Climbing Image Nudged Elastic Band)方法优化路径和计算锂离子迁移势垒[46]。需要指出的是,对于上述几种方法,随着计算精度的提高,所需要的运算时间会大幅增长,如对于单一结构的计算,半定量的键价和方法的计算量在分钟量级,而基于DFT的第一性原理计算则往往耗时数小时甚至数天。因此,将各种不同精度的方法合理组合,用于材料筛选的不同阶段,是有助于提高筛选效率的办法。
3.4电子结构
电池材料的能带结构也可以为材料筛选提供十分有价值的信息。电极材料中,过渡金属与氧的分波态密度的相对位置,决定了材料中电荷补偿的来源和氧的稳定性[1],从能态密度中还能定性预测电极与电解质界面的化学稳定性[47],此外电池的倍率性能也与电极的电子导电性有关[18]。电池中的固态电解质材料需要具有电子绝缘的特性,其电化学窗口的宽度与材料的能隙宽度有关[11]。因此电子结构的计算无论对电极材料还是电解质材料的筛选都十分必要。
电子结构的计算方法也可以分为半经验和第一性原理计算两类[48]。半经验方法常需要用到一些拟和参数,而基于密度泛函理论的第一性原理计算仅需要用到基本物理常数,在知道所需模拟体系的结构和组成的情况下,不需要其他经验的或拟和的可调参数,因此无论在计算过程还是在结果的精确度上都具有半经验方法不可比拟的优势。第一性原理从量子力学的基本方程出发,求解描述体系的波函数及相应的本征能量,并以此推导体系的各种性质,电子态的求解过程是一个自洽计算的过程,因此比半经验方法需要更多的计算时间[49]。因此有必要在材料筛选的不同阶段合理选择计算方法。
根据上述锂电池材料中各种性质的计算方法,我们针对电极材料和电解质材料分别需要满足的特性,构筑出筛选规则,对无机材料晶体结构数据库[50]里已有的结构进行高通量计算,并研究其性质与结构之间的关系。
4锂电池材料筛选的基本思路
在锂电池中,电极材料与电解质材料在性能上需要满足不同的要求。对于电极材料的研发,侧重于提高电极材料的比容量和提高电池的工作电压。对于固体电解质材料的研究,则希望找到离子电导率高,电化学窗口宽,并能与正负极稳定匹配的材料。此外所有的电池材料都需要具有较低的成本、无毒无放射性,减少对环境的污染,以期持续利用和发展。
在上述约束条件和筛选目标的指导下,我们首先考虑从无机材料晶体结构数据库(ICSD)[50]中,根据化合物的组成元素进行初步筛选,如图2所示,再进一步分别考虑它们作为电极或电解质材料的可能性。具体方案可以用图3表示。对于从ICSD中初步筛选出的化合物及其衍生物,首先考虑材料的储锂极限容量(优先考虑Li含量,其次考虑可转移电子数)。将计算出的理论容量按照140 mAh·g-1(LiCoO2材料的容量)为参考,如果大于这个参考值,则首先考虑其作为电极材料应用。接下来计算其脱/嵌Li电位,若电位合适,则继续计算脱Li后体积变化以及结构稳定性。如果脱Li后结构不稳定,我们则转而考虑其作为固体电解质。当电位、结构稳定性两个都符合要求,则继续计算离子扩散势垒等参数,并判断离子输运是否符合设置的约束条件。如果符合离子迁移势垒要求,则计算电子结构与晶格动力学参数,对前面的结果进行进一步修正,得到更为精确的数据。

图2 从ICSD数据库中初步筛选锂电池材料的方案Fig.2 Scheme of preliminary screening of lithium battery materials from ICSD database

图3 高通量计算筛选锂电池电极材料和固体电解质材料的流程Fig.3 Procedures of screening electrode materials and solid electrolytes using high-throughput calculation method
当开始计算的理论容量小于140 mAh·g-1时,我们直接考虑其为固体电解质材料的应用。我们首先判断其中是否含有可变价元素,如果有,则不再做其它考虑。如果不含可变价元素,我们则计算其电子结构,判断其电子绝缘特性,如果是电子导体或带隙很窄的半导体,那么这种材料也不能用做固体电解质,可以不再考虑。如果符合电子绝缘的条件,则考虑其分解电压。满足条件后,则进一步考虑Li离子在材料中的迁移势垒。如果满足迁移势垒条件,则进行晶格动力学参数修正。
在计算方法上,根据化合物中Li的含量以及可变价元素可能转移的电子数目来计算材料的理论储锂容量[38],采用热力学基本数据来计算电极反应的能量密度,并获得材料的平均嵌锂电位[38],对于缺少热力学数据的电极反应,采用DFT方法计算嵌锂电位[40],同时得到脱/嵌锂前后的体积变化。对于材料是否电子绝缘,以及对应的分解电位,也采用DFT计算来进行预测。对于锂离子传导的动力学性质,则采用半经验与DFT相结合的方法,先通过运算速度较快的键价和方法[44]对材料的离子输运性质进行初步筛选,将具有连通的离子通道并且活化能较低的化合物找出来,再选择更为精确的DFT计算,采用NEB方法[46]计算准确的迁移势垒,对于包含无序占位或部分占位的化合物,则考虑采用第一性原理的分子动力学(FPMD)[49]来进行计算。DFT计算主要采用VASP程序[37],基于键价和的离子通道计算则采用自行编写的BVpath进行。同时,我们根据图3的筛选流程编写了相应脚本,实现了对ICSD数据库中的晶体结构数据自动转化、对计算软件的自动调用、以及根据筛选条件将计算结果自动分类存储的过程。
5高通量计算筛选锂电池电极材料
提高锂电池能量密度的主要方法之一是提高电极材料的储锂容量。电极材料的储锂容量与可转移电子数、可转移离子数有关。目前几种重要的正极材料(LiCoO2、LiMn2O4、LiFePO4)以及与其类似组成的材料,其电子转移都是通过过渡金属的氧化还原来实现。根据等式(1)和(2)可以从热力学数据计算各种化学反应对应的能量密度。
根据化合物在25 ℃时的标准吉布斯自由焓数据以及密度数据[39],Zu等人[38]计算了1 172种化学反应所对应的能量密度。图4显示了部分锂电池体系的计算结果,当反应物的形成能较低而产物的形成能较高时,化学反应对应的能量密度较高。计算的所有体系中,Li/F2体系具有最高的能量密度,6 294 Wh·kg-1。其次是Li/O2体系,其能量密度为5 217 Wh·kg-1。除此之外,一些转化反应也显示出较高的能量密度。Zu等人[38]进一步计算了金属锂与各种物质发生转化反应的热力学平衡电压,部分结果如图5所示。对于二元过渡金属化合物与锂的转化反应,对于同种过渡金属,当转移电子数目相同时,热力学平衡电压值具有如下规律:氟化物>氧化物>硫化物>氮化物>磷化物,并且高氧化态的材料通常显示更高的热力学平衡电压值。
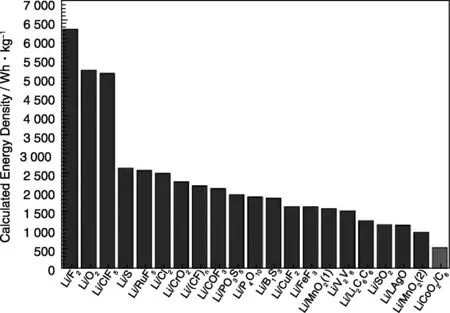
图4 根据能斯特方程计算的各种锂电池体系的能量密度[38]Fig.4 Calculated energy densities of different lithium battery systems according to the Nernst equation[38]

图5 计算的金属锂与各种二元过渡金属化合物发生转化反应的热力学平衡电压[38]Fig.5 Calculated thermodynamic equilibrium voltage of conversion reactions between binary transition metal compounds and lithium[38]
根据热力学数据计算的能量密度和热力学平衡电压,可有效用于电极材料(特别是正极材料)的初步筛选。以目前商业应用的正极材料LiCoO2为参考,将储锂能力高于此标准的化合物作为进一步筛选的候选材料。由于减少电极材料循环过程中的晶格体积变化对提高电池循环寿命至关重要,因此在接下来的研究中,将在高通量计算里引入第一性原理方法,对材料储锂前后的体积变化和热力学稳定性进行计算,以实现对上述候选材料的进一步筛选。
6高通量计算筛选锂电池固态电解质材料
从长远来看,开发全固态锂离子电池是解决目前锂电池发展瓶颈的一个思路。首先,全固态锂离子电池的安全性能非常好。当前锂离子电池采用液态电解液体系,采用的可燃的有机溶剂是电池体系中最大的安全隐患。而全固态锂离子电池采用固态电解质材料,安全问题可以从根源上一次性解决。其次,全固态锂离子电池可以采用金属Li为负极,其理论容量是锂电池体系的最高极限,而且其电位也是所有负极材料中最低的(这样可以提高电池体系的整体的电压)。因此,从总体上可以大大提高电池体系的能量密度。再次,由于负极材料采用了金属Li,这就给了正极材料更宽的选择——正极材料可以不作为Li源的提供者。
然而,要开发全固态锂离子电池体系,面临着许多的挑战,其中最关键的挑战在于开发出可以应用的固体电解质材料体系。固体电解质需要兼具高离子电导率、高稳定性、高机械强度等特点。目前的固体电解质体系有3类:①无机晶态固体电解质材料,包括石榴石(Garnets)结构材料[51]、金红石(Rutiles)结构材料[52]、沸石(Zeolites)类结构材料[53]等;②玻璃态的(Glassy)无定形态材料体系,这些材料通常都是Li的S化物和P化物(Li2S、P2S5、 Li3P7、Li7P3S11、 Li4P2S7等)[12];③ 聚合物(PEO)固态电解质材料体系[54]。这些材料体系都有其各自的优点和缺点,但目前尚没有获得可实际应用的固体电解质材料,因此发现新的固态电解质材料体系势在必行。
离子输运性质是决定固态电解质性能的最主要因素,因此计算材料的锂离子通道及活化能大小是进行电解质材料筛选的首要步骤。初步的筛选从计算精度较低但运算速度很快的键价和方法开始。BVpath程序结合了改进的键价和理论[44],计算流程如图6所示。从晶体结构数据库中读出化合物中的原子坐标、占据率和各原子的名义电荷,据此计算锂离子与阴离子之前的键能以及锂离子与阳离子之间的库伦排斥势,以二者之和来做等值面,找出能使锂离子通道联通的最小能量等值面,从而确定扩散路径和势垒。

图6 BVpath软件计算锂离子扩散路径及势垒的流程图Fig.6 Flow diagram of BVpath code based on bond valence method

图7 用键价和理论计算得到的Li2MnO3材料中的锂离子扩散路径:(a~c)为1.08 eV对应的等值面,显示了Li+在锂层内的迁移;(d~f)为1.55 eV对应的等值面,显示了Li+在过渡金属层与锂层之间的迁移Fig.7 The migration pathway of Li+ in Li2MnO3 calculated by Bond Valence method:(a~c)the isosurface of 1.08 eV indicates the migration of Li+ within the lithium layer and (d~f) the isosurface of 1.55 eV indicates the migration of a Li+ between the transition metal layer and lithium layer
图7是用键价和方法计算的Li2MnO3材料中的锂离子输运通道。以不同能量值画出的等值面显示了Li+在不同迁移路径上的能量势垒。从图(a)~(c)中可以看到,Li+在锂层之间的迁移的势垒为1.08 eV,低于Li+在过渡金属层和锂层之间的迁移势垒1.55 eV(图(d)~(f ))。键价和方法得到的锂离子输运路径与采用NEB方法的DFT计算结果[55]一致,二者都揭示出了沿着ab方向的层内传导和沿着c方向的层间传导两种路径;键价和方法计算得到的两种路径的势垒值由于受到计算方法的局限,在精度上无法与DFT计算比拟,但仍显示出层内传导比层间传导更易于发生,得到了与DFT计算结果一致的趋势。
通过对Li2MnO3计算结果的分析,表明了键价和方法在揭示晶体结构中可能存在的锂离子输运通道和半定量计算其迁移势垒的有效性。因此,这种方法可以更广泛的用于锂电池材料离子输运性质的研究。考虑到离子输运性质是固体电解质材料研发中需首要考虑的性质,我们编写了调用BVpath程序的高通量计算流程,对ICSD数据库中800余种含锂化合物的锂离子输运通道和迁移能量势垒进行了计算[56-57]。图8给出了7种分别属于不同晶系的化合物的计算结果。图中浅灰色的包络面是能量势垒值所对应的等值面,其中包裹的区域为Li+可能运动的区域。等值面所包裹的通道的最窄处为空间中的能量鞍点,是迁移路径中的能量最高点。根据键价和方法计算得到的离子通道和势垒值可以用于判断材料中锂离子运动趋势及其难易程度,从而用于从晶体结构数据库中初步筛选固体电解质。

图8 用键价和理论计算得到的7种材料的锂离子扩散通道与势垒:(a)Li2CO3,Ea=0.60 eV;(b) Li3B7O12,Ea=1.42 eV;(c) Li6Ge2O7,Ea=0.90 eV;(d) γ-LiAlO2,Ea=0.85 eV;(e) α-LiAlO2,Ea=1.89 eV;(f)LiAlSiO2 ,Ea=0.38 eV;(g) LiF ,Ea=1.07 eV[57]Fig.8 The pathways and energy barriers of Li+ migration for seven compounds:(a)Li2CO3,Ea=0.60 eV;(b) Li3B7O12,Ea=1.42 eV;(c) Li6Ge2O7,Ea=0.90 eV;(d) γ-LiAlO2,Ea=0.85 eV;(e) α-LiAlO2,Ea=1.89 eV;(f)LiAlSiO2 ,Ea=0.38 eV;(g) LiF ,Ea=1.07 eV[57]
在进一步的高通量计算中,引入第一性原理方法,计算各种备选材料的电子结构,获得能隙宽度的数据,以此估算固体电解质材料的电化学窗口宽度,作为下一步的筛选条件。这些进一步的高通量计算和材料筛选工作正在进行中,预期将为锂电池中固体电解质材料的发现提供有价值的参考。
7结语
应用高通量计算方法探索和设计锂电池新材料,是未来材料科学研究和开发的一条重要途径。借助无机材料晶体结构数据库中现有的和衍生的结构,设定合理的限制条件,有望筛选出下一代锂电池的备选材料。作者团队自主编写了可实现晶体结构选择、输入文件生成、VASP软件调用、计算结果分类存储的自动化计算流程,并编写了基于键价和方法的锂离子输运计算软件BVpath,同时也实现了高通量计算过程对该软件的自动调用。
从材料研发的角度,这种方法有可能从现有晶体结构中找到或构建出新晶体结构,并评估新结构存在的可能性以及作为锂电池材料的性能,更快找到高性能、高安全性的正极、负极与电解质材料,促进锂电池产业的发展,加速锂电池材料的开发过程。
从基础研究的角度,高通量计算得到的大量数据,将有助于理解与锂电池有关的现象,如结构与离子扩散、储锂容量之间的本质关系。计算得到的晶体结构、电子结构和各种性质,与现有的实验数据汇集到一起,将形成一个庞大的数据库,为材料学家及物理学家提供全面而宝贵的数据参考。进一步借助统计方法进行数据挖掘,还有可能找到决定材料性质的因素,如相关的结构单元或是通过统计方法定义的关联函数等,从而找到背后的物理原因。
可以预见在未来几年内,应用高通量计算方法,结合实验研究,将大大加速我们对锂电池材料中各种问题的理解,揭示材料的结构-性能关系及内在物理规律,缩短材料从研发到应用的全过程,为开发全新电池材料乃至电池体系提供有力支持。
参考文献References
[1]Goodenough J B, Kim Y. Challenges for Rechargeable Li Batteries[J].ChemistryofMaterials, 2010 (22): 587-603.
[2]Tarascon J M, Armand M. Issues and Challenges Facing Rechargeable Lithium Batteries[J].Nature, 2001 (414): 359-367.
[3]Karden E, Ploumen S, Fricke B,etal. Energy Storage Devices for Future Hybrid Electric Vehicles[J].JPowerSources, 2007 (168): 2-11
[4]Dunn B, Kamath H, Tarascon J M. Electrical Energy Storage for the Grid: A Battery of Choices[J].Science, 2011 (334): 928-935
[5]Li H, Wang Z X, Chen L Q,etal. Research on Advanced Materials for Li-Ion Batteries[J].AdvancedMaterials, 2009 (21): 4 593-4 607.
[6]Chen Y, Wang G X, Konstantinov K,etal. Synthesis and Characterization of LiCoxMnyNi1-x-yO2as a Cathode Material for Secondary Lithium Batteries[J].JournalofPowerSources, 2003 (119): 184-188.
[7]Palacin M R, Chabre Y, Dupont L,etal. On the Origin of the 3.3 and 4.5 V Steps Observed in LiMn2O4-Based Spinels[J].JournalofTheElectrochemicalSociety, 2000 (147): 845-853.
[8]Hu Y, Kong W, Li H,etal. Experimental and Theoretical Studies on Reduction Mechanism of Vinyl Ethylene Carbonate on Graphite Anode for Lithium Ion Batteries[J].ElectrochemistryCommunications, 2004 (6): 126-131.
[9]He Y, Yu X, Wang Y,etal. Alumina-Coated Patterned Amorphous Silicon as the Anode for a Lithium-Ion Battery with High Coulombic Efficiency[J].AdvancedMaterials, 2011 (23): 4 938-4 941.
[10]Xu K. Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries[J].ChemicalReviews, 2004 (104): 4 303-4 417.
[11]Quartarone E, Mustarelli P. Electrolytes for Solid-State Lithium Rechargeable Batteries: Recent Advances and Perspectives[J].ChemicalSocietyReviews, 2011 (40): 2 525-2 540.
[12]Mizuno F, Hayashi A, Tadanaga K,etal. New, Highly Ion-Conductive Crystals Precipitated from Li2S-P2S5Glasses[J].AdvancedMaterials, 2005 (17): 918-921.
[13]Kamaya N, Homma K, Kamakawa Y,etal. A Lithium Superionic Conductor[J].NatureMaterials, 2011 (10): 682-686.
[14]Mo Y, Ong S P, Ceder G,etal. First Principles Study of the Li10GeP2S12Lithium Super Ionic Conductor Material[J].ChemistryofMaterials, 2012 (24): 15-17.
[15]Lu J, Li L, Park J B,etal. Aprotic and Aqueous Li-O2Batteries[J].ChemicalReviews, 2014 (114): 5 611-5 640.
[16]Bruce P G, Freunberger S A, Hardwick L J,etal. Li-O2and Li-S Batteries with High Energy Storage[J].NatureMaterials, 2012 (11): 19-29.
[17]Liu Y L, Wang R, Lyu Y C,etal.Rechargeable Li/CO2-O2(2:1) Battery and Li/CO2Battery[J].EnergyEnvironmentScience, 2014 (7): 677-681.
[18]Shi S Q, Liu L J, Ouyang C Y,etal. Enhancement of Electronic Conductivity of LiFePO4by Cr Doping and Its Identification by First-Principles Calculations[J].PhysicalReviewB, 2003 (68): 195 108.
[19]Ouyang C Y, Shi S Q, Wang Z X,etal. First-Principles Study of Li Ion Diffusion in LiFePO4[J].PhysicalReviewB, 2004 (69): 104 303.
[20]Ouyang C Y, Shi S Q, Wang Z X,etal. Ab Initio Molecular-Dynamics Studies on LixMn2O4as Cathode Material for Lithium Secondary Batteries[J].EurophysicsLetters, 2004 (67): 28-34.
[21]Wei Y J, Xu X G, Wang C Z,etal. Electronic Structure of Cubic Li(Fe0.1Mn1.9)O4Studied with Mössbauer Spectroscopy and First-Principles Calculation[J].AppliedPhysicsLetters, 2003 (83): 1 791-1 793.
[22]Van der Ven A, Ceder G, Asta M,etal. First-Principles Theory of Ionic Diffusion with Nondilute Carriers[J].PhysicalReviewB, 2001(64): 184 307.
[23]Ouyang C Y, Zhong Z Y, Lei M S,etal. Ab Initio Studies of Structure and Electronic Properties of Li4Ti5O12Spinel[J].ElectrochemistryCommunications, 2007(9): 1 107-1 112.
[24]Ouyang C Y, Zeng X M,ljivancanin Ž,etal. Oxidation States of Mn Atoms at Clean and Al2O3-Covered LiMn2O4(001) Surfaces[J].JournalofPhysicalChemistryC,2010(114): 4 756-4 759.
[25]Ten Bosch J R, Grody W W. Keeping Up with the Next Generation[J].TheJournalofMolecularDiagnostics, 2008 (10): 484-492.
[26]Tucker T, Marra M, Friedman J M. Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine[J].TheAmericanJournalofHumanGenetics, 2009 (85): 142-154.
[27]Howe D, Costanzo M, Fey P,etal. Big Data: The Future of Biocuration[J].Nature,2008 (455): 47-50.

[29]Andersson M P, Bligaard T, Kustov A,etal. Toward Computational Screening in Heterogeneous Catalysis: Pareto-Optimal Methanation Catalysts[J].JournalofCatalysis, 2006 (239): 501-506.
[30]Curtarolo S, Setyawan W, Wang S D,etal. AFLOWLIB.ORG: A Distributed Materials Properties Repository from High-Throughput Ab Initio Calculations[J].ComputationalMaterialsScience, 2012 (58): 227-235.
[31]Setyawan W, Gaumé R M, Lam S,etal. High-Throughput Combinatorial Database of Electronic Band Structures for Inorganic Scintillator Materials[J].ACSCombinatorialScience, 2011 (13): 382-390.
[32]Yang K, Setyawan W, Wang S,etal. A Search Model for Topological Insulators with High-Throughput Robustness Descriptors[J].NatureMaterials, 2012 (11): 614-619.
[33]Jain A, Hautier G, Moore C J,etal. A High-Throughput Infrastructure for Density Functional Theory Calculations[J].ComputationalMaterialsScience, 2011 (50): 2 295-2 310.
[34]Hautier G, Jain A, Chen H,etal. Novel Mixed Polyanions Lithium-Ion Battery Cathode Materials Predicted by High-Throughput Ab Initio Computations[J].JournalofMaterialsChemistry, 2011 (21): 17 147-17 153.
[35]Hautier G, Jain A, Ong S P,etal.Phosphates as Lithium-Ion Battery Cathodes: An Evaluation Based on High-Throughput Ab Initio Calculations[J].ChemistryofMaterials, 2011 (23): 3 495-3 508.
[36]Fujimura K, Seko A, Koyama K,etal. Accelerated Materials Design of Lithium Superionic Conductors Based on First-Principles Calculations and Machine Learning Algorithms[J].AdvancedEnergyMaterials, 2013 (3): 980-985.
[37]Kresse G, Furthmuller J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set[J].ComputionalMaterialsScience, 1996 (6): 15-50.
[38]Zu C X, Li H. Thermodynamic Analysis on Energy Densities of Batteries[J].EnergyEnvironmentalScience, 2011 (4): 2 614-2 624.
[39]David R.CRCHandbookofChemistryandPhysics, 90thedn[M].Boca Raton:CRC Press, 2009.
[40]Ceder G, Chiang Y-M, Sadoway D R,etal. Identification of Cathode Materials for Lithium Batteries Guided by First-Principles Calculations[J].Nature, 1998 (392): 694-696.
[41]Anurova N A, Blatov V A. Analysis of Ion-Migration Paths in Inorganic Frameworks by Means of Tilings and Voronoi-Dirichlet Partition: A Comparison[J].ActaCrystallographicaSectionB:StructuralScience, 2009 (65): 426-434.
[42]Nuspl G, Takeuchi T, Weiss A,etal. Lithium Ion Migration Pathways in LiTi2(PO4)3and Related Materials[J].JournalofAppliedPhysics, 1999 (86): 5 484-5 491.
[43]Filso M O, Turner M J, Gibbs G V,etal. Visualizing Lithium-Ion Migration Pathways in Battery Materials[J].Chemistry-AEuropeanJournal, 2013 (19): 15 535-15 544.
[44]Brown I D. Recent Developments in the Methods and Applications of the Bond Valence Mode[J].ChemicalReview, 2009 (109): 6 858-6 866.
[45]Adams S, Rao R P. High Power Lithium Ion Battery Materials by Computational Design[J].PhysicaStatusSolidiA, 2011 (208): 1 746-1 753.
[46]Sheppard D, Xiao P, Chemelewski W,etal. A Generalized Solid-State Nudged Elastic Band Method[J].JournalofChemicalPhysics, 2012,136(7): 074 103.
[47]Lepley N D, Holzwarth N A W, Du Y A. Structure, Li+Mobilities, and Interfacial Properties of Solid Electrolytes Li3PS4and Li3PO4from First Principles[J].PhysicalReviewB, 2013,88(10): 104 103.
[48]Singleton J.BandTheoryandElectronicPropertiesofSolids[M].Oxford University Press, 2001.
[49]Martin R M.ElectronicStructure:BasicTheoryandPracticalMethods[M]. Cambridge University Press, 2004.
[50]Inorganic Crystal Structure Database, ICSD. Karlsruhe: Fachinformationszentrum,2008.
[51]Murugan R, Thangadurai, Weppner W. Fast Lithium Ion Conduction in Garnet-Type Li7La3Zr2O12[J].AngewandteChemieInternationalEdition, 2007 (46): 7 778-7 781.
[52]Yates D E, Healy T W. Titanium Dioxide-Electrolyte Interface[J].JournaloftheChemicalSociety,FaradayTransactions1: Physical Chemistry in Condensed Phases, 1980 (76): 9-18.
[53]Noto V D. Zeolitic Inorganic-Organic Polymer Electrolyte Based on Oligo(Ethylene Glycol) 600 K2PdCl4and K3Co(CN)6[J].JournalofPhyscialChemistryB, 2000 (104): 10 116-10 125.
[54]Jacob M M E, Hackett E, Giannelis E P. From Nanocomposite to Nanogel Polymer Electrolytes[J].JournalofMaterialsChemistry, 2002,1(1): 1-5.
[55]Xiao R, Li H, Chen L Q. Density Functional Investigation on Li2MnO3[J].ChemistryofMaterials, 2012 (24): 4 242-4 251.
[56]Gao J, Chu G, He M,etal. Screening Possible Solid Electrolytes by Calculating the Conduction Pathways Using Bond Valence Method[J].ScienceChina:Physics,MechanicsandAstronomy, 2014,57(8): 1 526-1 535.
[57]Zhang Shu(张舒), Wang Shaofei(王少飞), Ling Shigang(凌仕刚),etal. 锂离子电池基础科学问题(X)—— 全固态锂离子电池[J].EnergyStorageScienceandTechnology(储能科学与技术), 2014,3(4): 376-394.
(编辑盖少飞)
Application of High-Throughput Calculations for ScreeningLithium Battery Materials
LING Shigang1, GAO Jian1, CHU Geng1, HUANG Jie1, XIAO Ruijuan1,
OUYANG Chuying2, LI Hong1, CHEN Liquan1
(1.Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China)
(2.Jiangxi Normal University, Nanchang 330027, China)
Abstract:Advanced cell technology is the world’s leading competition in the next ten years. Lithium batteries are the key to the success of portable electronic products, electrical vehicle and the best choices for the energy storage devices of wind and solar power because of their high voltage, high specific energy density, rapid recharge capability, and wide working temperature range. The development of the next generation lithium battery materials is necessary to achieve higher energy density, better safety, and faster charge and discharge speed. The development of new materials is possible to be accelerated by high-throughput calculations and reasonable screening procedures. In this work, using automated calculation flow designed by ourselves, the initial screening of electrode matreials and solid electrolytes is performed from the Inorganic Crystal Structure Database according to the application requirments. The calculation results and experimental data integrate into a large database, which is hopeful to reveal the realtionship between structure and property, thus shortening the process of materials from discovering to application and providing support for the development of new battery materials and even new battery systems.
Key words:lithium battery material; high-throughput calculation; thermodynamic calculation; bond valence method
中图分类号:O646.2+1
文献标识码:A
文章编号:1674-3962(2015)04-0272-10
DOI:10.7502/j.issn.1674-3962.2015.04.03
通讯作者:肖睿娟,女,1980年生,副研究员,Email: rjxiao@aphy.iphy.ac.cn
基金项目:国家自然科学基金(11234013,51325206,51172274);北京市科委重点项目;科技部973项目(2012CB932900)
收稿日期:2014-09-04
