
庆大霉素生物合成基因genB4的研究
2015-02-25 01:08温淑平林强洪文荣
延边大学学报(自然科学版) 2015年4期
温淑平, 林强, 洪文荣
( 福州大学 生物科学与工程学院, 福建 福州 350116 )
庆大霉素生物合成基因genB4的研究
温淑平,林强,洪文荣*
( 福州大学 生物科学与工程学院, 福建 福州 350116 )
摘要:以庆大霉素生物合成基因簇为模版,构建了genB4基因阻断质粒pGB403.经接合转移导入绛红色小单孢菌G1008,得到一株genB4基因缺失工程菌GB4408.对菌株GB4408的发酵产物进行了TLC、HPLC和MS分析,结果表明GB4408不再合成庆大霉素C族组分,而是积累中间代谢产物G418、西索霉素和威大霉素,阻断了从西索霉素到庆大霉素C1a的转化以及从威大霉素到庆大霉素C2的转化,这可能是genB4基因参与了庆大霉素绛红糖胺C4′和C5′的双脱氢作用.据此推论,在菌株GbK(△genK)基础上敲除了genB4基因,并成功获得一株主产西索霉素的工程菌GbKB4,进一步验证了genB4的功能. 绛红色小单孢菌;genB4; 西索霉素 Q93
文献标识码:A
庆大霉素是由小单孢菌发酵液中分离得到的一组广谱抗生素,包含庆大霉素C1、C1a和C2等组分.在化学结构上,庆大霉素由2-脱氧链霉胺(2-DOS)、绛红糖胺和加洛糖胺3部分组成.研究[1-3]发现,庆大霉素的生物合成是从X2开始,分为2条路线(图1):一条经JI-20A、西索霉素合成庆大霉素C1a;另一条经G418、JI-20B、威大霉素合成庆大霉素C2,并最终合成庆大霉素C1.2条合成路线的酶促反应只对绛红糖胺进行修饰.近年来,不同菌株的庆大霉素生物合成基因簇陆续被公布出来.F.Kudo等[1]通过与妥布霉素、卡那霉素生物合成基因簇比较,推测genB4为B型的氨基转移酶基因.Guo J.H.等[4]通过基因失活研究,推测基因genB4与绛红糖胺C3′,4′-双脱羟基有关.本实验组[5]也对genB4基因进行了失活,结果产生了带有新化合物的复杂组分的产物,但是否由genB4单基因失活而导致这一结果仍有待进一步验证;因此,有必要重新对genB4基因进行敲除.
本文通过基因框内敲除方法,重新设计引物,构建同源重组质粒,阻断绛红小单孢菌G1008中genB4(1 338 bp)的表达,并借助分析代谢物的变化,进一步研究genB4基因在庆大霉素生物合成过程中所起到的作用.

图1 庆大霉素生物合成途径
1材料与方法
1.1 材料
1) 质粒与菌株.克隆载体使用pMD19-T(日本TaKaRa),用温敏型自杀质粒pKC1139[6]作为大肠杆菌-链霉菌属间穿梭载体.用E.coliTop10
作为质粒克隆宿主菌,用短小芽孢杆菌〔CMCC(B) 63202〕作为抗生素微生物检定菌,用E.coliET12567(pUZ8002)[7]作为大肠杆菌—链霉菌属间接合转移供体菌,用绛红色小单孢菌G1008和GbK(△genK)作为接合转移受体菌.
2) 培养基与抗生素.绛红色小单孢菌斜面培养基、种子培养基、发酵培养基以及孢子预萌发培养基参照文献[8].抗生素微生物检定培养基Ⅰ参照2010版《中华人民共和国药典》,大肠杆菌培养使用LB培养基,各培养基根据需要添加相应的抗生素.本研究使用的抗生素作用浓度分别为:氨苄青霉素100 μg/mL,安普霉素50 μg/mL,氯霉素25 μg/mL,卡那霉素25 μg/mL,萘啶酮酸25 μg/mL.
3) 工具酶与试剂.限制性核酸内切酶、T4 DNA Ligase、Taq DNA聚合酶、DNA Marker和Proteinase K(日本TaKaRa公司);DNA凝胶回收试剂盒、RNase A酶和溶菌酶(上海生工生物工程有限公司);其他常规试剂见参考文献[9].
1.2 方法
1)引物设计.以基因库(Genebank)公布的庆大霉素生物合成基因簇(登录号:JQ975418)为模板,利用生物信息学软件Vector NTI11.5设计两对引物扩增genB4基因上下游序列作为同源交换臂:引物P1/P2用于扩增上游交换臂B41,引物P3/P4用于扩增下游交换臂B42.设计引物P5/P6用于genB4基因阻断突变株的验证.所有引物由上海生工生物工程股份有限公司合成,引物位置与扩增片段长度如图3所示,引物序列及其限制性酶切位点见表1.
2) DNA分子操作.PCR、质粒DNA抽提、大肠杆菌感受态细胞的制备、酶切、酶连和转化等常规分子克隆操作参照文献[10].绛红色小单孢菌基因组DNA的提取和接合转移参照文献[11].

表1 研究genB4所用引物
3)工程菌发酵及产物提取分析.绛红色小单孢菌发酵及产物提取方法参照文献[8],生物效价测定参照2010版《中华人民共和国药典》.提取的产品采用硅胶GF254薄层层析(TLC)和HPLC进行初步检测,TLC展开剂为氯仿-甲醇-氨水(1∶1∶1,体积比),混合均匀后静置5 min,展开;HPLC参照2010版《中华人民共和国药典》,组分的精确测定采用质谱法.
2结果与讨论
2.1 重组质粒pGB403的构建
提取绛红色小单孢菌G1008基因组DNA,并以其为模板,PCR扩增genB4上下游交换臂B41和B42.将这两条交换臂分别克隆至pMD19-T载体,得到中间质粒pGB401和pGB402.质粒pGB401用Hind Ⅲ和EcoRⅠ酶切,回收1 963 bp片段;质粒pGB402用Hind Ⅲ和XbaⅠ酶切,回收2 019 bp片段;pKC1139用XbaⅠ和EcoRⅠ酶切,回收6 446 bp片段.将以上3个片段酶连,酶连样转化Top10感受态细胞,筛选阳性克隆子,最终获得重组质粒pGB403.用EcoRⅠ和XbaⅠ对pGB403进行酶切,酶切样经电泳检测(图2)有两条带与理论值(6 446 bp和3 994 bp)相符,最后通过测序验证,证明质粒pGB403为正确质粒.
2.2 GB4408菌株的构建
将重组质粒pGB403转化E.coliET12567(pUZ8002)得到接合转移供体菌E.coliET12567(pGB403/pUZ8002),绛红色小单孢菌G1008的孢子作为接合转移受体菌.按照接合转移方法,将孢子悬液与供体菌混合,稀释涂布于平板培养基,于37 ℃培养约20 h后,再用含安普霉素和萘啶酸的水溶液覆盖,继续培养直至在平板上长出接合子.

图2 质粒pGB403酶切电泳检测图(1:PGB403,XbaⅠ/EcoRⅠ; 2:DL 5 000 Marker)
选取一株长势较好的接合子,提取基因组DNA,用P5/P6引物进行PCR扩增验证.如果得到两条片段,分别为1 241 bp和512 bp,那么说明其为单交换工程菌.PCR产物电泳结果与预期相符(图3B),这表明重组质粒pGB403已成功整合到绛红色小单孢菌G1008,即单交换突变株菌,将其命名为小单孢菌GB41.
由于单交换菌株具有同源性极高的片段,不稳定,容易发生2次同源重组,获得genB4基因缺失双交换菌株或回复突变菌株,质粒pGB403与G1008同源重组示意图如图3A所示.

1:DL 5 000 Marker; 2:G1008(P5/P6); 3:GB41(P5/P6); 4:GB4408(P5/P6)图3 GB4408基因组DNA PCR产物电泳图
将单交换菌株GB41于37 ℃、不含抗生素的斜面上松弛培养3代,然后分离单菌落,通过单菌落影印点板方法进行筛选.经筛选,从408株菌株中获得2株在抗性平板上不生长,而在无药平板上正常生长的单菌落.为验证其是否为目的工程菌,选取其中一株长势良好的菌株,提取其基因组DNA,然后利用双交换鉴定引物P5/P6进行PCR验证.如果电泳只检测到512 bp的片段,而未检测到1 241 bp的片段,那么该菌株即为双交换工程菌.电泳检测结果表明所挑选的菌株为双交换工程菌,将其命名为GB4408,如图3所示.将PCR产物测序后,其结果进一步说明GB4408工程菌缺失了genB4基因729 bp.
2.3 GB4408菌株的代谢产物分析
GB4408的发酵液经树脂吸附、乙醇沉淀等处理之后,获得粗制样品.将样品进行TLC分析,结果如图4.图中主要有3个斑点,与标准品相比,未合成庆大霉素C1、C1a、C2等庆大霉素C族组分,转而合成其他庆大霉素中间体.

TLC:1 G1008, 2 GB4408图4 GB4408菌株代谢产物的TLC和HPLC分析
按照《中华人民共和国药典》[12],对GB4408代谢产物进行HPLC分析,检测结果见图4.庆大霉素标准品各组分的保留时间分别为:C1(5.98 min),C1a(14.58 min),C2a(18.57 min),C2(21.05 min).GB4408的粗制样品中的3个主峰,保留时间分别为7.68、18.72、20.12 min,这与庆大霉素标准品的保留时间无一相符.由此进一步表明GB4408的代谢产物没有庆大霉素C族组分.
为精确确定GB4408代谢产物的组分,采用MS分析,结果如图5所示.图中主要离子峰有4个,分别为448.3(西索霉素)、462.3(威大霉素)、476.3(威大霉素C6′位甲基化产物)、497.3(G418).231.6和249.1分别为威大霉素和G418的双电荷峰,322.2为碎片峰.这说明工程菌GB4408不再合成庆大霉素C族组分,而是积累中间代谢产物G418、西索霉素和威大霉素,阻断了西索霉素到庆大霉素C1a和威大霉素到庆大霉素C2的转化,据此推测genB4参与庆大霉素绛红糖胺C4′和C5′的双脱氢作用.

图5 GB4408菌株代谢产物的MS分析
2.4 GbKB4工程菌的构建
绛红小单孢菌GbK菌高产庆大霉素C1a,且genK基因失活,阻断了从庆大霉素X2至G418的转化.根据对genB4基因功能的推测,若在菌株GbK上继续敲除genB4基因,则有望获得西索霉素的工程菌.
利用上述方法,将供体菌ET12567/(pGB403/ pUZ8002)与GbK(△genK)的孢子进行接合转移,7 d后长出2株接合子,挑取长势良好的一株(命名为GbKB4-1).将其转接至小单孢菌种子培养基,松弛培养4代后开始分离单菌落,并影印到含50 μ/mL安普霉素和不含抗生素的平板上,筛选后获得工程菌GbKB4,提取其基因组DNA,用引物P5/P6进行PCR验证,电泳检测见图6.

1: GB4408(P5/P6); 2: DL 5 000 Marker; 3: GbK(P5/P6)图6 GbKB4基因组DNA PCR产物电泳图
2.5 工程菌GbKB4发酵及代谢产物分析
按照同样的方法,对工程菌GbKB4进行发酵及代谢产物的提取,组分分析采用HPLC,见图7.以西索霉素标准品作对照,按照庆大霉素的流动相来分析,西索霉素标准品出峰的保留时间为18.64 min.GbKB4粗制样品有2个主峰,保留时间分别为6.21 min和18.63 min.其中后者与标准品的出峰时间大致相符,由此初步确认其为西索霉素.

图7 GbKB4菌株代谢产物的HPLC分析
精确组分分析采用质谱检测,如图8所示.GbKB4工程菌离子峰448[M+H]+、470[M+Na]+、486[M+K]+,它们与西索霉素分子量同质.离子峰483[M+H]+,它与中间代谢产物庆大霉素X2同质.242[M+H]+为庆大霉素X2双电荷峰,其他峰为碎片峰或杂质峰.这表明GbKB4菌株中敲除genB4基因,阻断了西索霉素转化庆大霉素C1a,由此获得了一株主产西索霉素工程菌,同时也验证了genB4基因的功能.
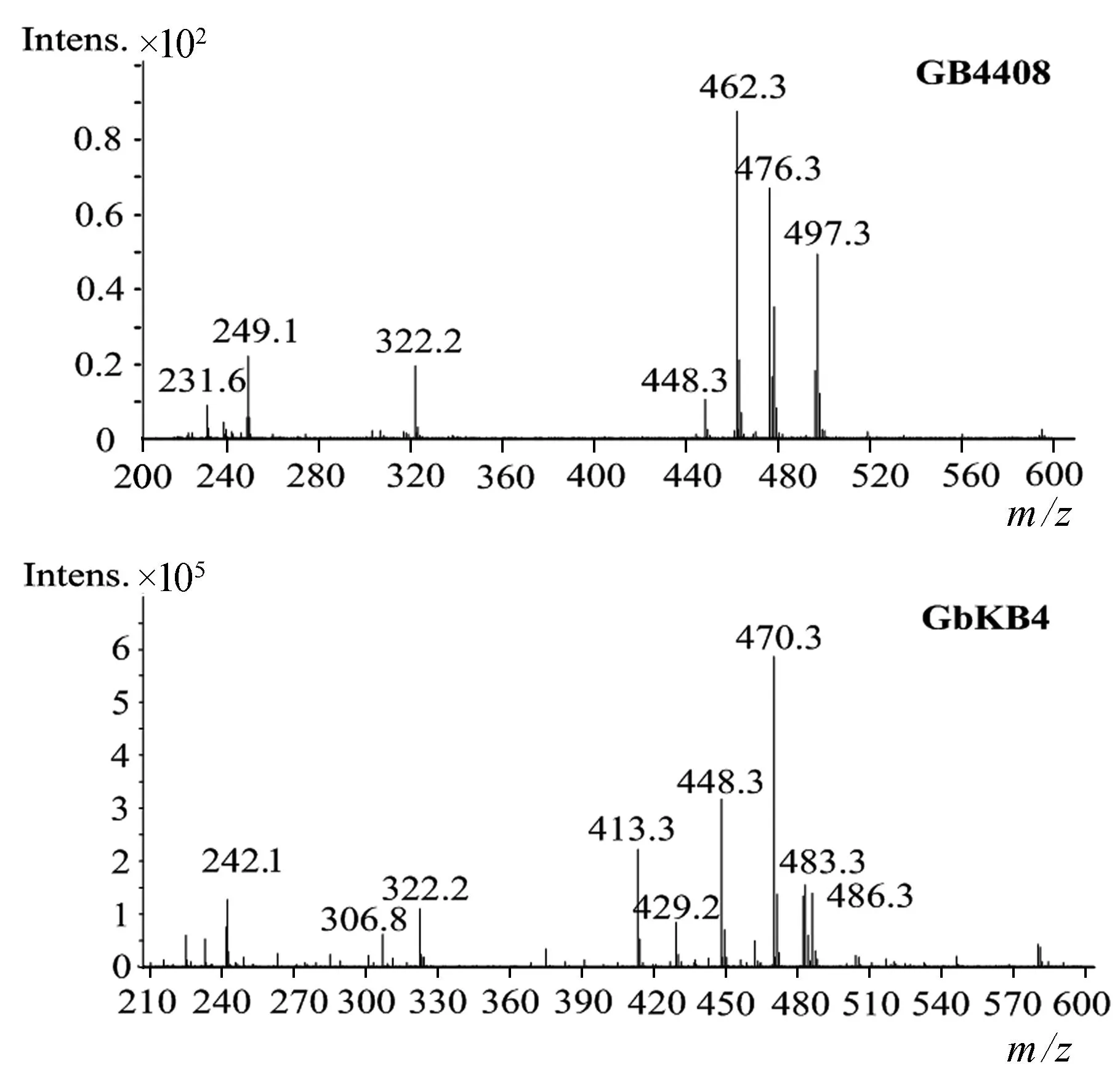
图8 GbKB4菌株代谢产物的MS分析
3结论
本文在绛红色小单孢菌G1008中,对庆大霉素生物合成基因genB4进行框内敲除,获得工程菌GB4408.对其代谢产物分析,发现其不再合成庆大霉素C组分,而是积累中间代谢产物G418、西索霉素和威大霉素.genB4基因的失活,阻断了从西索霉素到庆大霉素C1a的转化以及从威大霉素到庆大霉素C2的转化,推测genB4基因参与了庆大霉素绛红糖胺C4′和C5′的双脱氢作用.由此推测,在genK基因阻断工程菌GbK的基础上敲除genB4基因后获得genK和genB4双基因失活的工程菌GbKB4.将GbKB4发酵后,提取其代谢产物,经HPLC和MS分析结果表明,工程菌GbKB4主要积累中间体西索霉素和庆大霉素X2.与工程菌GB4408相比,工程菌GbKB4不再合成G418和威大霉素,因此本文成功地构建了一株主产西索霉素的工程菌,同时也进一步验证了genB4基因的功能.
参考文献:
[1]Kudo F, Eguchi. Biosynthetic genes for aminoglycoside Antibiotics[J]. The Journal of Antibiotics, 2009,62:471-481.
[2]UnwinJ,StandageS,AlexanderD,etal.GeneclusterinMicromonospra echinosporaATCC15835forthebiosynthesisofthegentamicinCComplex[J].TheJournalofAntibiotics, 2004,57(7):436-445.
[3]AboshanabK.Geneticstudiesonthebiosynthesisofthemajoraminoglycosideantibiotics.Dissertation[D].Wuppertal:BergischeWuppertalUniversity, 2005.
[4]GuoJH,HuangFL,HuangC,etal.Specificityandpromiscuityatthebranchpointingentamicinbiosynthesis[J].Chemistry&Biology, 2014,21:608-618.
[5]洪文荣,张熠,张书祖,等.庆大霉素生物合成基因研究进展[C]//十二届全国抗生素学术会议论文集.四川:中国抗生素杂志社,2013:452-466.
[6]FlettF,MersiniasV,SmithCP.HighefficiencyintergenericconjugaltransferofplasmidDNAfromEscherichiacolitomethylDNA-restrictingstreptomycetes [J].FEMSMicrobiologyLetters, 1997,155(2):223-229.
[7]ParanthamanS,DharmalingamK.IntergenericconjugationinStreptomyces peucetiusandStreptomycessp.strainC5:chromosomalintegrationandexpressionofrecombinantplasmidscarryingthechiCgene[J].AppliedandEnvironmentalMicrobiology, 2003,69(1):84-91.
[8]HongWR,YanLB.IdentificationofgntK,agenerequiredforthemethylationofpurpurosamineC-6′ingentamicinbiosynthesis[J].JournalofGeneralandAppliedMicrobiology, 2012,58(5):349-356.
[9]TobiasK,BibbM,MarkJB,etal.PracticalStreptomycesGenetics[M].Norwich:TheJohnInnesFoundation, 2000.
[10]BiermanM,LoganR,ObrienK,etal.PlasmidcloningvectorsfortheconjugaltransferofDNAfromEscherichia colitoStreptomycesspp.[J].Gene, 1992,116(1):43-49.
[11]严凌斌,洪文荣,方志锴,等.绛红色小单孢菌G1008接合转移体系的构建[J].中国抗生素杂志,2011,36(12):899-904.
[12]国家药典委员会.中华人民共和国药典[M].2部.北京:中国医药科技出版社,2010:976-977.
Research ofgenB4 in gentamicin biosynthesis gene cluster
WEN Shuping,LIN Qiang,HONG Wenrong*
(CollegeofBiologicalScienceandTechnology,FuzhouUniversity,Fuzhou350116,China)
Abstract:A recombinant plasmid pGB403 was constructed with gentamicin biosynthesis gene cluster as the template to study the function ofgenB4. Then, the plasmid pGB403 was transformed intoMicromonosporapurpureaG1008 by conjugation. A disruptgenB4 was obtained, i.e.,emqineeringstrain(GB4408). Its ferment extract was analysised by TLC, HPLC and MS. The GB4408 mainly produced accumulated intermediate G418, sisomicin and verdamycin instead of gentamicin C compounds. The metabolic flux from sisomicin to gentamicin C1a and from verdamycin to getamicin C2 were blocked. This result indicated thatgenB4 might be responsible for the dehydrogenation at C4′- C5′of purpurosamine. Based on the speculation,genB4 was distrupted in the strain GbK(△genK), and the strain GbKB4 mainly produced sisomicin was successful constructed, which further verified the function ofgenB4.
Key words:Micromonosporapurpurea;genB4; sisomicin
文章编号:1004-4353(2015)04-0313-05
基金项目:国家自然科学基金资助项目(31070093);国家“重大新药创制”科技重大专项项目(2012ZX09201101-008)
收稿日期:2015-09-23*通信作者: 洪文荣(1956—),男,博士,教授,研究方向为微生物制药.
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
山东农业科学(2022年2期)2022-03-12
浙江农业学报(2021年1期)2021-01-28
三农资讯半月报(2020年11期)2020-06-21
东北农业大学学报(2020年3期)2020-05-14
农民致富之友(2017年13期)2018-01-27
中西医结合心血管病电子杂志(2016年14期)2016-11-17
中国市场(2016年23期)2016-07-05
