
新方法合成三芳基铋、三芳基锑化合物
2015-02-24 03:34:04温运明邓向军
暨南大学学报(自然科学与医学版) 2015年3期
温运明, 邓向军, 唐 渝
(暨南大学生命科学技术学院化学系,广东广州510632)
新方法合成三芳基铋、三芳基锑化合物
温运明, 邓向军, 唐 渝
(暨南大学生命科学技术学院化学系,广东广州510632)
在氮气条件下,建立了一个简单有效的合成对称的三芳基铋、三芳基锑化合物方法:1.0倍量的三氯化铋、溴代芳烃和镁屑各4.5倍量,无配体和催化剂,在四氢呋喃溶液中,温度为65℃的条件下反应10 h.并在空气条件下,将合成的一系列三芳基锑化合物应用于催化安息香氧化生成二苯基乙二酮的反应,根据反应时间与收率两个因素,讨论了取代基电子及位阻效应对其催化效果的影响.
合成; 三芳基铋化合物; 三芳基锑化合物; 催化
有机铋、锑化合物在有机合成、生物药物、催化应用等方面有着重要的应用[1-5],其中三芳基铋和锑化合物作为C-C键形成的重要的有机金属试剂得到了广泛关注[6-7].它们不仅可以作为催化剂,在有机合成中三芳基铋化合物还可作为配体[8].无水卤化物与有机镁或锂试剂反应是制备三芳基铋和三芳基锑最经典的方法[9].但是有机金属试剂(Mg,Li)需在无水条件下预先制备,有时还需在低温下进行反应(-70℃,Li),且收率较低[10].有机锌试剂和三氯化铋反应也能制备三芳基锑,但是底物限定为邻位有吸电子基的芳基卤代物而且需要长时间反应(18 h)[11].邻位有吸电子基的碘代芳烃与铋粉在铜粉和CuI条件下[12]也可以制备三芳基铋,但是反应操作比较复杂.四芳基硼酸钠与醋酸铋(III)进行芳基化也可以得到三芳基铋[13].目前的制备方法还存在反应复杂、反应时间长以及原材料昂贵等缺点,因此,寻找更符合绿色化学[14-15]这一概念且操作简单的三方基铋或锑的合成方法仍然具有重要意义.
通过实验研究,发现三芳基铋可以通过一锅法制备,方法简单,溶剂四氢呋喃(tetrahydrofuran)无需除水处理,无需配体和催化剂.该方法的反应条件为:在氮气条件下,在分析纯四氢呋喃(15 mL)中,用芳基溴代物、镁各4.5倍量与三氯化铋或者三氯化锑1倍量在65℃反应(图1).
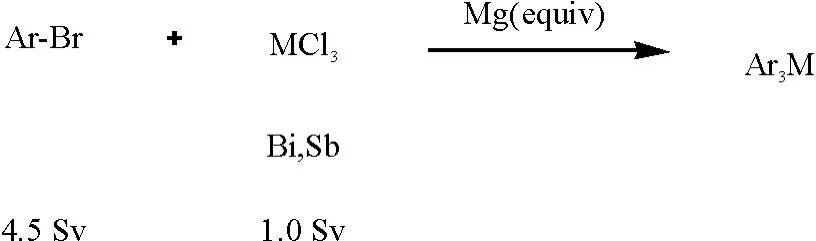
图1 芳香卤代烃和三氯化铋反应Fig.1 reactions of alkyl halides with BiCl3/SbCl3
1 实验部分
1.1 三芳基铋和三芳基锑的制备通法
取11.25 mmol芳基溴代物、11.25 mmol打磨后的镁屑,在氮气氛围下、加入到15 mL四氢呋喃中,再加入2.5 mmol的BiCl3或SbCl3(在手套箱里称量),65℃下反应10 h.反应完成后冷却到室温、抽滤,滤渣用少量氯仿洗涤3次,与滤液合并,加无水硫酸镁干燥,再过滤,滤液旋转蒸发得到粗产物,取少量粗产物进行过薄层层析(展开剂为石油醚)大板分离,获得分离收率,所有产物均为已知化合物,经1HNMR和质谱结构鉴定并与文献对比确定结构.
1.2 三芳基锑催化空气氧化安息香生成二苯基乙二酮
[15]方法,将2 mmol安息香、0.2 mmol三芳基锑加入到8 mL CH2Cl2,通入空气,在30℃条件下反应.薄层层析(展开剂为:乙酸乙酯∶石油醚=1∶5)跟踪反应,直到原料消失,记录反应时间.反应结束后过滤、旋蒸得到粗产物,取少量通过薄层层析(展开剂为石油醚)大板分离,得到最终的分离产率.
三苯基铋:无色晶体;MS∶m/z=440(M+),363,287,77.IR(KBr):3 044,1 944,1 566,1 472,1 425,1 326,1 299,1 183,1 155,1 013,996,723,693,450 cm-1;1HNMR(300 MHz,CDCl3):δ=7.76,(d,J=6.6,6H),δ=7.34-7.43(m,9H)ppm;数据和文献[13]报道相同.
三-(对甲基苯基)铋:无色晶体;MS:m/z=391,300,209,91.2,77.2;1HNMR(300 MHz,CDCl3):δ7.36(d,J=7.8,6H),7.16(d,J=7.8,6H),2.38(s,9H)ppm;IR(KBr):3 005,2 915,1 905,1 584,1 486,1 437,1 385,1 308,1 261,1 207,1 182,1 051,1 011,851,788,562 cm-1;数据和文献[13]报道相同.
三-(邻甲基苯基)铋:无色晶体;MS(EI):m/z=482(M+),391,300,299,209,91;1HNMR(CDCl3):δ7.56-7.58(d,J=7.50,3H),7.35-7.37(d,J=7.50,3H),7.28(dt,J1=1.2 and J2=7.4,3H),7.07(t,J=7.4,3H),2.47(s,9H)ppm,IR(KBr):2 963,1 444,1 199,1 154,1 113,747,535;数据和文献[13]报道相同.
三-(2,4-二甲基苯基)铋:无色晶体;MS(EI):m/z=419.1,314.1,209,105.2,77.1;1HNMR(CDCl3):δ7.42(d,J=7.5 3H),6.88(s,J=7.8,3H),7.03(d,J=7.9,3H),2.40 s,9H),2.30(s,9H)ppm;IR(KBr):3 006,2 965,2 918,2 918,2 851,1 593,1 466,1 439,1 228,1 110,810,552 cm-1;数据和文献[13]报道相同.
三-(对甲基苯基)锑:无色晶体;MS(EI):m/z=442.3(M+),335.2,228.1,121.1,91.0,77.0;1H NMR(CDCl3):δ3.80(d,9H),6.88(d,6H,J=8.6 Hz),7.73(d,6H,J=8.6 Hz)ppm;数据和文献[16]报道相同.
三-(间氟苯基)锑:无色晶体;MS(EI):m/z=407.0(M+),310.9,215.9,95.0,75.0;1HNMR(CDCl3):δ7.04-7.35(m,12H)ppm;IR(KBr):3 049,2 969,2 851,1 578,1 566,1 469,1 399,1 229,1 031,989,858,825,797,694,648,558 cm-1;数据和文献[17]报道相同.
2 结果与讨论
2.1 最优条件探索
在表1中我们研究了催化剂和配体对反应的影响,FeCl3、AlCl3、PdCl2等路易斯酸催化反应得到的收率分别为20%、44%、31%(表1,Entries 1-3).使用CuI、Cu2O、CuO和CuBr2等一些铜催化剂获得的反应收率分别为41%、50%、34%、48%(表1,Entries 4-7),有一定提高.考虑到配体与催化剂的搭配,选择了在CuBr2催化下,分别加入含N、P、O的三乙胺、三苯基膦、乙二醇二甲醚、四甲基乙二胺等配体,获得的反应收率分别分18%、20%、12%、16%(表1,Entries 8-11),收率的下降表明加入配体不利于反应的进行.考虑BiCl3也可以作为催化剂使用,考察了不加其他金属催化剂和配体的反应,收率能达到64%(表1,Entry 12),在此基础上加入四丁基溴化铵及其他的一些配体进行反应,收率没有提高(表1,Entries 13-17).
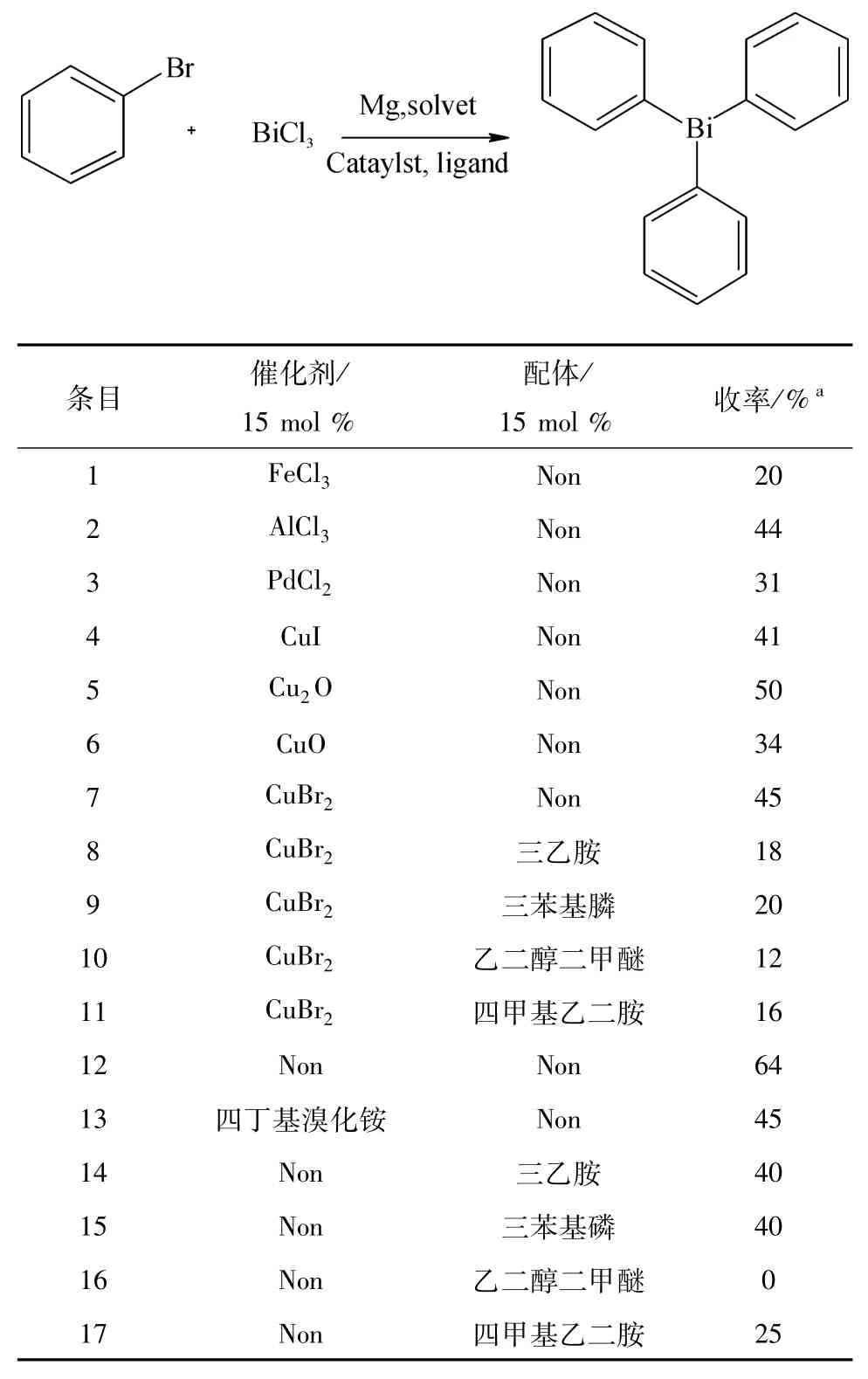
表1 催化剂和配体的影响Table 1 Effects of catalysts,ligands on the reaction
在上面实验结果的基础上,进一步考察了反应温度的影响,65℃反应得到的收率最高为64%(表2,Entry 2),反应温度降低到40℃或升高到80℃都不利于反应的进行,收率分别为55%(表2,Entry 1)或51%(表2,Entry 3).同时在65oC的条件下通入氮气进行反应,收率提高到了78%.
在表3中,增加反应时间到10 h的收率从78%(表3,Entry 1)提高到84%(表3,Entry 2),继续延长反应时间到12 h,收率为85%(表3,Entry3).接着对溴苯或镁的量进行了讨论,在10h相同反应时间的条件下,从4.5倍量减少溴苯或镁的用量,收率从84%(表3,Entry 2)明显下降到了61%(表3,Entry 4),而继续增加其用量到5倍,收率变化不大为84%(表3,Entry 5).综合考虑,溴苯和镁4.5倍量,反应10 h最佳.
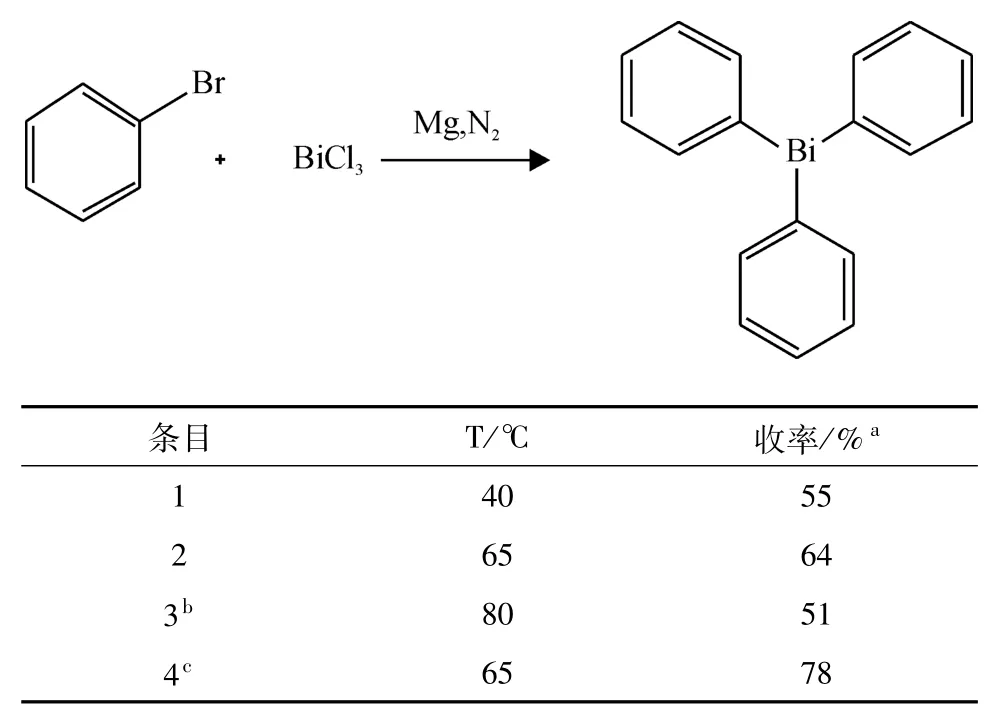
表2 温度和通入氮气的影响Table 2 Effects of temperature and nitrogen on the reaction
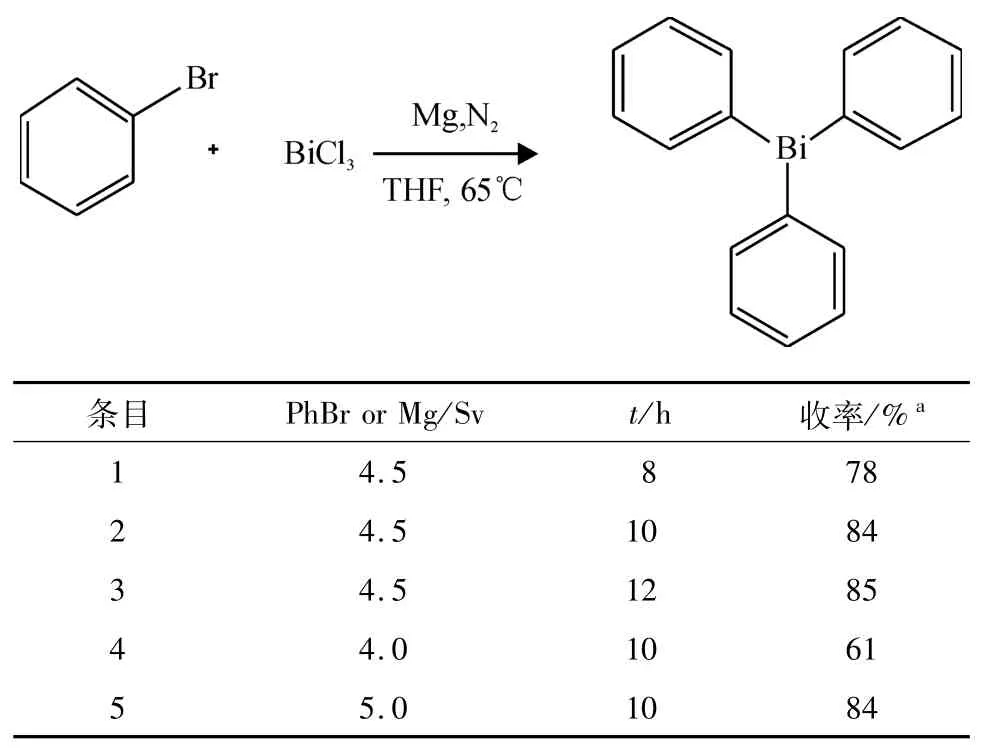
表3 时间和溴苯的用量的影响Table 3 Effects of PhBr or Mg amount and reacting time on the reaction
2.2 不同的芳香卤代烃和三氯化铋/三氯化锑反应
在以上最优的反应条件下,三氯化铋和锑与不同三芳基溴代物的反应,结果见表4.在碘、溴和氯苯的相关反应中,溴苯获得的收率最高,为84%(Bi)和80%(Sb)(表4,Entry 1),虽然碘苯的反应活性比溴苯高,但其生成产物的收率却下降到60%左右(表4,Entry 1),氯苯由于活性低,几乎没有产物生成(表4,Entry 1).对位和邻位甲基溴苯没有表现出明显的位阻和给电子效应,其对应的产物收率接近约70%(表4,Entries2,3),相对于没有取代基的溴苯有所下降.而邻甲基三芳基锑产物的收率则比对甲基的高分别为72%和86%(表4,Entries 2,3),从三芳基锑的80%收率看(表4,Entry 4),2,4-二甲基溴苯在反应中也基本呈现出相似的规律,其生成的三芳基铋与对甲氧基溴苯一样都只获得中等收率分别为50%和45%(表4,Entries 4,5),尤其是后者比其邻位产物的收率67%(表4,Entry 6)还要低.对甲氧基和邻甲氧基溴苯对应的三芳基锑的收率则呈现出正常的对甲氧基给电子效应,收率提高到90%,邻甲氧基的位阻效应影响收率为80%(表4,Entry 6).间甲氧基溴苯分别得到了40%和88%收率的三芳基铋和锑的产物(表4,Entry 7),其与对位甲氧基一样使三芳基铋收率下降,这有可能是甲氧基中的氧原子与金属铋有配位作用而影响了反应,当邻位有溴原子时的这种配位作用会减弱,因此没有降低反应收率.综合考虑,仍可以认为甲氧基溴苯所获得的反应结果没有表现出明显的甲氧基在间位时弱的吸电子作用,这和氯取代时的结果一致(表4,Entries 8,9).对和邻氯溴苯所得三方基铋的收率分别为50%、65%,与前面的规律相似,三芳基锑的收率都接近85%(表4,Entries8,9),没有明显表现出吸弱电子基的影响和位阻效应.虽然通常情况下溴代萘的反应活性相对较低,但三芳基铋和锑仍获得了较好的约60%的收率(表4,Entry 10).3-氟溴苯和2-溴噻吩的产率因反应活性较低其生成的三芳基铋只有20%和16%的较低收率,但生成的三芳基锑的收率则相对较好,分别为55%和68%(表4,Entries 11,12).总的来说,三芳基锑大部分都能获得80%以上的较好收率,效果较好.
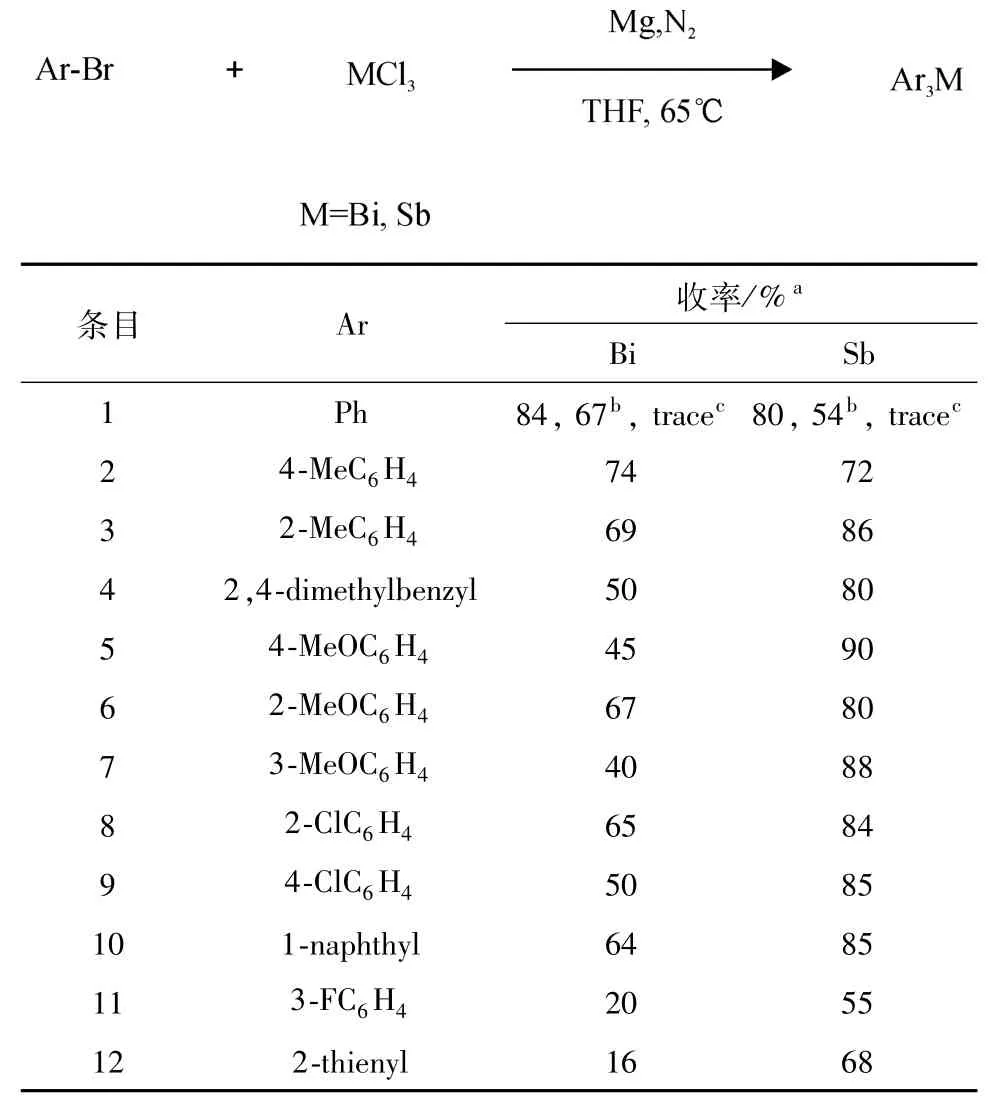
表4 芳香卤代烃和三氯化铋或三氯化锑反应Table 4 Reactions of aryl bromides with BiCl3or SbCl3
2.3 三芳基锑的催化作用
α-二酮苯偶酰是一种重要的有机中间体,如用作光敏材料和合成试剂.氧化α-酮醇是合成对苯偶酰最高效、实用的方法[14],其中三芳基锑是一种有效的催化剂,能催化氧气将二芳基-α-酮醇氧化成相应的α-二酮,使用其他氮族试剂如三苯基磷、砷或铋等不会反应[15].由于用新方法能有效地合成三芳基锑,为探讨所合成三芳基锑的应用效果,我们在文献[17]基础上将其用于催化氧化安息香生成二苯基乙二酮的反应中(图2),结果见表5,其中甲基、甲氧基、间氟及邻氯取代三苯基、三噻吩基和三萘基锑在文献[17]中都没有催化反应的收率数据(表5,Entries 2-8,10-12).

图2 三芳基锑催化空气氧化安息香生成二苯基乙二酮Fig.2 Oxidation of benzoin into benzil with air catalyzed by triarylstibines
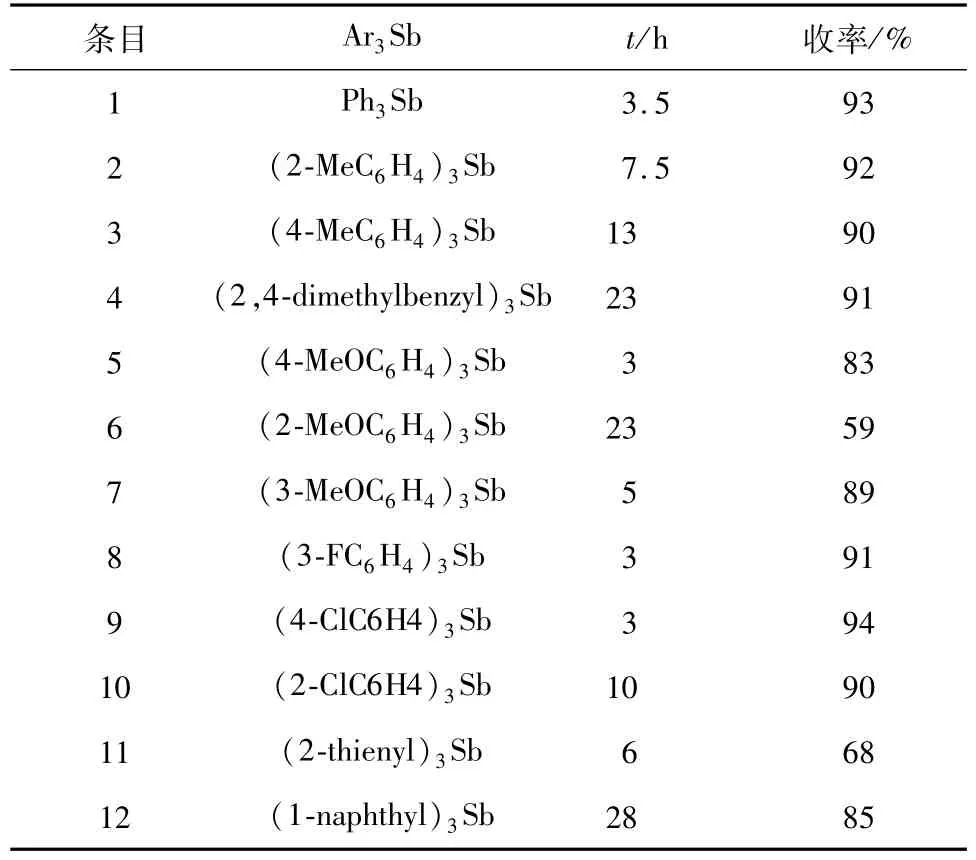
表5 三芳基锑催化空气氧化安息香生成二苯基乙二酮Table 5 Oxidation of benzoin into benzil with air catalyzed by triarylstibines
研究发现,三芳基锑中,苯基、对或邻甲基取代的苯基都取得了90%及以上的高收率(表5,Entries 1-4),甲基的给电子效应和位阻效应没有影响反应收率,但邻位的位阻效应使反应时间大大增长(表5,Entries 3-4).邻、间和对位取代的甲氧基也呈现相似的规律,但由于甲氧基相对较强的给电子作用及位阻效应,邻甲氧基三苯基锑催化的反应收率出现了明显下降,而对位和间位取代的甲氧基三苯基锑只是略降低了反应收率(表5,Entries 5-7).吸电子的F和Cl对反应较有利,在3 h反应后,产物的收率已经能达到90%以上(表5,Entries8-9),邻位取代的氯获得90%的反应收率也只是使反应时间延长到10 h(表5,Entry 10),从效果来看,仍然比邻位取代的甲基略好,这些规律与文献[17]报道一致,说明我们制备的三芳基锑已达到市售产品的效果.三噻吩锑和三萘锑也有不错的催化效果(表5,Entries 11-12).
3 结论
通过对反应条件的探索,获得了制备三苯基铋的最优化条件,即在通入氮气的条件下,4.5倍的溴苯和镁、1倍量的BiCl3,在15 mL四氢呋喃中,65℃一锅反应10 h.在此条件下,制备了不同结构的三芳基铋和锑(40%~92%).与以前的方法比较,该方法简单,THF无需处理,无配体和催化剂,大多数产物有良好的收率.在空气条件下,将合成的三芳基锑化合物应用于催化安息香氧化生成二苯基乙二酮,获得了与文献[17]一致的规律,达到了市售产品的效果.
[参考文献]
[1] NICHOLAS M LEONARD,LAURE C WIELAND,RAM S MOHAN,et al.Applications of bismuth(III)compounds in organic synthesis[J].Tetrahedron,2002,58(42):8373-8397.
[2] GREGORY I ELLIOTT,JOSEPH P KPNOPELSKI.Arylation with organolead and organobismuth reagents[J],Tetrahedron,2001,57(27):5683-5705.
[3] JAY P PARRISH,TERRY V HUGHES,HWANG I,et al.Establishing the parabolic relationship between reactivity and activity for derivatives and analogues of the duocarmycin and alkylation subunits[J].J Am.Chem.Soc,2004,126(1):80-81.
[4] YOSHIHIRO MATANO,HAZUMI NOMURA,HISANAGA T,et al.Diverse structures and remarkable oxidizing ability of triarylbismuthane oxides.Comparative study on the ostructure and reactivity of a series of triarylpnictogen oxides[J].Organometallics,2004,23(23):5471-5480.
[5] MADDDLI L N RAO,DEBASIS BANERJEE,DHANORKARR J,et al.Pd(0)-catalyzed couplings using bromide and chloride derivatives of Baylis-Hillman adducts with triarylbismuths as atom-efficientmulti-coupling nucleophiles[J].Tetrahedron,2010,66(20):3623-3632.
[6] TAKASHI NISHIKATA,YASUNORI YAMAMOTO,GRIDNEV ID,et al.Enantioselective 1,4-Addition of Ar3Bi,[ArBF3]K,and ArSiF3 to enones catalyzed by a dicationic palladium(II)-chiraphos or dipamp complex[J].Organometallics,2005,24(21):5025-5032.
[7] MADDALI L N RAO,DEBASIS BANERJEE,DEEPAK N JADHAV.Palladium catalyzed atom-efficient crosscoupling reactions of triarylbismuths with aryl iodides and aryl triflates[J].Tetrahedron Letters,2007,48(38):6644-6647.
[8] PAVEL ARSENYAN,MARTINS IKAUNIEKS,SERGEY BELYAKOV.Stille coupling approaches for the synthesis of 8-aryl guanines[J].Tetrahedron Letters,2007,48(6):961-964.
[9] MADDALI L N RAO,SOMNATH,DEEPAK N JADHAV.Pd-catalyzed synthesis of a-aryl ketones through couplings of a-arylacetyl chlorides with triarylbismuths as multi-coupling nucleophiles[J].Tetrahedron Letters,2009,50(45):6133-6138.
[10]LIX W,LORERTH J,MASSA W,et al.Synthesis and characterization of aryl bismuth compounds using 2,4,6-triphenylphenyl as a bulky ligand[J].Journal of Organometallic Chemistry,1995,485(1):141-147.
[11]CHRISTOPHE PICHON,MURIEL PIPELIER,URGIN K,et al.Advanced preparation of functionalized triarylbismuths and triheteroaryl-bismuths:new scope and alternatives[J].Tetrahedron Letters,2012,53(15):1894 -1896.
[12]MIKA URANO,SNINOBU WADA,HITOMI SUZUKI.A novel dry route to ortho-functionalized triarylbismuthanes that are difficult to access by conventional wet routes[J].Chem.Commun,2003(10):1202-1203.
[13]VITALIE STAVILA,JOHN THURSTON.A new methodology for synthesis of aryl bismuth compounds:arylation of bismuth(III)carboxylates by sodium tetraarylborate salts[J].Organometallics,2007,26(27):6864-6866.
[14]陈 兵,龙俞霖,杨 骏,等.微波加热促进功能化离子液体催化合成柠檬酸三辛酯[J].暨南大学学报:自然科学与医学版,2012,5(33):490-495.
[15]高培培,陈河如.抗癌药物泰克地那林的合成工艺优化[J].暨南大学学报:自然科学与医学版,2012,3(33):286-288.
[16]MADDALI L N RAO,DEEPAK N,DASGUPTA P,et al.Pd-catalyzed tandem chemoselective synthesis of 2-arylbenzofurans using threefold arylating triarylbismuth reagents[J].Eur.J.Org.Chem,2013,2013(4):781-788.
[17]SHUJI YASUIKE,YOSHIHITO KISHI,KAWARA S,et al.Catalytic action of triarylstibanes:oxidation of benzoins into benzyls using triarylstibanes under an aerobic condition[J].Chem.Pharm.Bull,2005,53(4):425 -427.
[责任编辑:刘蔚绥]
A simple and efficient procedure for the synthesis of symmetrical triarylbismuth and triarylstine
WEN Yunming, DENG Xiangjun, TANG Yu
(Department of Chemistry,College of Life Science and Technology,Jinan University,Guangzhou 510632,China)
Under nitrogen atmosphere,a simple and efficient procedure for the synthesis of symmetrical triarylbismuth or triarylstine has been developed by reactiing aryl or alkyl bromides and magnesium(4.5 equiv)with BiCl3or SbCl3(1.0 equiv)in the absence of catalyst and ligand in THF at65℃for10 h.Under air atmosphere,a series of synthesized triarylstibines has been applied in the catalyzed oxidative reaction of benzoin into benzyl.The effects of electronic and steric hindrance of substituents on the reaction have been discussed according to the reaction times and yields.
synthesis; triarylbismuth; triarylstibine; catalysis
O621.3
A
1000-9965(2015)03-0212-06
10.11778/j.jdxb.2015.03.004
2015-01-05
国家大学生创新性试验计划项目(101055920);暨南大学本科生科技创新工程项目(cx10067)
唐 渝(1968-),教授,博士,研究方向:有机合成方法学.Tel:020-85222786;E-mail:tytang@jnu.edu.cn
猜你喜欢
农药科学与管理(2019年8期)2019-11-23 08:04:44
中国麻业科学(2018年6期)2018-04-09 11:22:26
低碳世界(2016年29期)2016-11-09 01:35:36
当代化工研究(2016年7期)2016-03-20 16:21:50
合成化学(2015年2期)2016-01-17 09:04:21
化工进展(2015年6期)2015-11-13 00:27:23
中国塑料(2015年10期)2015-10-14 01:13:13
化学工业与工程(2015年1期)2015-02-10 03:01:33
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年7期)2014-02-28 17:32:28
