
国际生物信息学研究的可视化分析
2015-02-23 07:58李延晖
生物信息学 2015年4期
游 鸽,李延晖,刘 向
(华中师范大学 信息管理学院 , 武汉 430079)
国际生物信息学研究的可视化分析
游鸽,李延晖*,刘向
(华中师范大学 信息管理学院 , 武汉 430079)
摘要:利用当前主流的信息可视化分析软件CiteSpace对2005~2014年间SCI收录的生物信息学的5种高影响力外文期刊所刊载论文的题录数据进行统计和可视化分析,绘制该领域的关键词共现、膨胀词共现、经典文献共现、高被引文献共现和关键节点文献共现的网络可视化图谱,试图揭示生物信息学领域的研究热点、研究前沿以及知识基础,以期帮助研究人员了解该领域在国际范围内的研究态势。
关键词:生物信息学;CiteSpace;信息可视化;知识图谱;研究前沿 是作者对文章核心研究内容的精炼,学科领域里高频次出现的关键词可被视为该领域里的研究热点[13]。在CiteSpace软件界面选择关键路径探测算法(Pathfinder)——该算法经过模型运算来剪切网络中大部分不重要的节点关联,只保留最重要的节点关联,同时让所有的节点均保持不动,以便从最大程度上将原网络简化为一个最小值网络[14, 15];网络节点选为关键词;数据抽取对象设为top30;设置TimeScaling的值为1(即将2005~2014年分成10个时段进行处理)。运行CiteSpace,得到生物信息学关键词共现图,见图1。
“生物信息学”是英文“Bioinformatics”的中文译名,1991年美国学者Lim在其发表的文章首次使用该词[1]。生物信息学是包含了生物信息的获取、处理、储存、分析和解释等在内的所有方面的一门交叉学科,它是综合数学、计算机科学和生物学的各种工具进行研究,目的在于了解和阐明大量生物信息学数据所包含的生物意义[2]。
进入21世纪,生物信息学相关出版物井喷式增加,俨然成为当下研究热点领域之一。为了厘清生物信息学研究的发展脉络,尽快获悉国际同行的研究动向,国外多位学者对生物信息学领域的研究趋势进行了相应的研究,比如:Ouzounis C A 运用定性分析的方法对生物信息学的早期发展阶段做了回顾[3]。Patra, SK 对PubMed数据库中主题为生物信息学的研究文献进行了计量分析,试图揭示该领域的演变历程和发展趋势[4]。Perez-Iratxeta C 对生物信息学的演化和发展趋势做了研究,并将生物信息学定性为具有惊人增长动力的新兴学科[5]。Glanzel, W 对生物信息学领域的核心文献的出版活动和引文影响力进行了比较分析[6]。Song M对PubMed数据库中2000至2011年生物信息学领域的文献与引文进行了计量分析,并指出了该领域最有成效的作者、机构、国家以及最流行的主题词[7]。近些年,国内也有多位学者从定性或定量多个视角对生物信息学领域的研究热点进行了相关研究,其中,王玉梅采用科学计量学和统计学方法对CBMdisk 生物医学文献数据库、中国期刊网2002年以前国内正式发表的生物信息学文献和首届中国生物信息学会议论文进行了统计分析,试图发现该领域的演化路径与发展趋势[8]。宋茂海利用共词分析的方法对生物信息学的关键词进行聚类分析,试图探讨该研究领域的学科分类和热点内容[9]。种乐熹和胡德华对发表在Nucleic Acids Research 期刊上有关生物信息学软件研究的文献做了可视化分析,试图揭示了该领域的重要研究力量、知识基础与研究热点[10]。生物信息界进行的类似回顾研究或对研究前沿和热点的探测,不仅能较好的展示该学科领域某一时间内的变化规律,还有助于该领域的学者们更快、更好地了解本研究领域的研究重点与发展趋势。
以美国科学情报研究所编辑出版的2014年自然科学版期刊引证报告为依据,选取5种高影响力外文期刊在2005~2014年所刊载研究论文的题录数据作为研究样本,利用信息可视化软件——CiteSpace,结合文献计量的相关方法,从多方面对生物信息学的研究状况、研究热点和前沿进行可视化分析,以期有益于我国生物信息学的研究工作。
1 数据与方法
1.1 数据来源
期刊引用报告依据来自 ISI Web of Science 平台中引文数据,是一种独特的多学科期刊评价工具。本文选取2014年自然科学版期刊引证报告中生物信息学专业目录下综合影响力较高和相关性较强的5种期刊作为数据来源,期刊名依次为Bioinformatics、BMC Bioinformatics、Briefings in Bioinformatics、PLoS Computational Biology、Nature Genetics,检索数据库选定为Web of Science TM核心合集,检索期限设定为2005~2014,出版类型设定为article,共计检索到18 789条符合条件的数据记录(检索时间:2015年11月4日),下载的方式设定为全纪录(包含引用的参考文献)。
1.2 研究工具和方法
引文分析可视化是信息可视化的重要分支,在处理完海量的引文数据之后,利用信息可视化技术使人们更直观地观察浏览和理解信息,进而找到数据中隐藏的规律和模式[11]。当前开展引文分析多应用统计学中的一些工具,如SPSS、Pajek等,但是其可视化的效果较为单调,信息可视化工具CiteSpace软件[12]正好弥补上述不足,并且它在时序分析和热点凸显上具有显著的特征和优势。
采用CiteSpace软件,将18 789篇以全记录形式保存的纯文本题录数据导入信息可视化软件进行相应的分析处理,绘制关键词共现、膨胀词共现、经典文献共现、高被引文献共现、关键节点文献共现的网络可视化图谱,借此来揭示国际生物信息学领域的研究状况和发展动态。
2研究热点与研究前沿
2.1 研究热点探测

图1 生物信息学领域主要研究热点知识图谱Fig.1 Knowledge map of hot domains on bioinformatics
图1共有84个节点,20条连接线,图中每个圆形节点代表关键词,节点及其标签大小与词汇出现的频次成正比,较大的节点可视为近十年国际生物信息学领域主要研究热点主题,表1是近十年国际生物信息学领域研究热点词频统计。通过词藻聚类和主题分析得到国际生物信息学的研究热点主要集中在基因组与遗传学研究、蛋白组学研究、细胞与分子生物学研究、基因的数据挖掘分析、生物系统建模与仿真等五大领域,依次对应图1中#A、#B、#C、#D、#E五个子聚类。
2.2 研究前沿分析
研究某学科领域的研究前沿对该学科领域研究人员具有重要意义,可使研究者及时准确地把握学科研究前沿和最新演化动态,还可预测学科发展的方向和未来需进一步研究的热点问题[16]。探测研究前沿可利用CiteSpace的膨胀词探测算法,通过考察词频的时间分布,将其中频次变化率高的名词短语从主题词中探测出来,依靠词频的变动趋势,而不仅仅是频次的高低,来确定学科领域的研究前沿[17]。在软件界面选择膨胀词探测算法;网络节点选为膨胀词;收据抽取对象设为top50;设置TimeScaling的值为1。运行CiteSpace绘制出近十年国际生物信息学领域研究前沿与趋势知识图谱,见图2。
如图2所示:该共引网络是由427个节点、73条连线组成,图中突变名词短语频次最高的是酵母(Saccharomyces-cerevisiae)、其次是序列比对 (Sequence-alignment)、蛋白质序列(Protein-sequence)、氨基酸(Amino-acids)、补充信息(Supplementary-information)、人类基因组(Human-genome)、基因表达谱(Gene-expression-profiles)、比值比(Odds-ratio)、序列相似性(Sequence-similarity)等。从图2可看出国际生物信息学前沿主要有功能基因组与比较基因组学、蛋白质结构比对与预测、分子进化分析、生物计算等领域:

图2 生物信息学领域研究前沿与趋势知识图谱Fig.2 Knowledge map of frontiers and trends on bioinformatics
(1)表征功能基因组与比较基因组学作为生物信息学前沿的突变名词短语包括序列比对 (Sequence-alignment)、人类基因组(Human-genome)、基因表达谱(Gene-expression-profiles)、对比分析(Comparative-analysis)、对照基因组(Reference-genome)、多序列比对(Multiple-sequence-alignments)、基因组序列(Genomic-sequences)等。
(2)表征蛋白质结构比对与预测作为生物信息学前沿的突变名词短语包括蛋白质序列(Protein-sequences)、氨基酸(Amino-acids)、氨基酸序列(Amino-acid-sequence)、蛋白质值数据银行(Protein-data-bank)、相互作用的蛋白质(Interacting-proteins)、蛋白质家族(Protein-family)等。
(3)分子进化分析作为生物信息学前沿的突变名词短语包括酵母(Saccharomyces-cerevisiae)、补充信息(Supplementary-information)、比值比(Odds-ratio)、分子动力模拟(Molecular-dynamics-simulations)、分子生物学(Molecular-biology)、拟南芥(Arabidopsis-thaliana)、共变异(Common-variants)、分子机制(Molecular-mechanism)等。
(4)表征生物计算作为生物信息学前沿的突变名词短语包括计算模型(Computational-model)、支持向量机(Support-vector-machines)、强大工具(Powerful-tool)、芯片技术(Microarray-technology)、统计效率(Statistical-power)、微阵列数据分析(Microarray-data-analysis)等。
另外,从图2中还可以发现,近十年来生物信息学领域研究前沿还有如下内容:生物途径(Biological-pathways)、系统发育树(Phylogenetic-tree )、Web应用(Web-application)、生命科学(Life-sciences)、印迹分析(Blot-analysis)、人类疾病(Human-diseases)等。
3知识基础分析
通过对知识基础进行分析,可使研究者更好地了解生物信息学的发展脉络和研究基础。知识基础有助于进一步指出研究前沿的本质,从文献计量学的角度来看,引文形成了研究前沿,被引文献生成了知识基础[18]。开展学科领域知识基础的分析包括对早期奠基性文献分析以及对一组共被引频次和中心性都比较高的关键文献分析[19]。通过绘制近十年国际生物信息学领域文献共被引网络,分别进行时间、被引频次和中心性三方面的分析,进而明确生物信息学领域的知识基础。
3.1 早期经典文献
早期奠基性文献是科学领域后期发展的坚实基础,通过对近十年国际生物信息学领域被引文献进行时间分析可得出该领域的早期奠基性文献。设置参数运行CiteSpace并选择时间线(Timeline)视图,得到近十年国际生物信息学领域早期奠基性文献时间序列知识图谱,见图3。

图3 国际生物信息学早期奠基性文献时间序列知识图谱Fig.3 Time line knowledge map of early foundational literatures on international bioinformatics
图3显示发表于1970~1999年间的9篇早期奠基性文献。1970年,Needleman et al.发表了A general method applicable to the search for similarities in the amino acid sequence of two proteins一文,该文首次引入了一种利用计算机寻找两种蛋白质的氨基酸序列之间的相似性的自适应方法,该方法可以判断蛋白质之间是否存在显著同源性,并由此来追溯它们的进化发展历程[20]。1977年,Dempster et al在Maximum Likelihood from Incomplete Data via the EM Algorithm一文中提出了一种计算最大似然估计广泛适用的算法——EM算法[21],后来该算法及其改进型被广泛用于生物计算中。1981年,Smith et al.发表了Identification of common molecular subsequences一文,该文指出如果我们假设连续子序列没有内部缺失或插入(片段),那么解决最大的同源套之间的子序列的序列识别这个问题将变得很简单[22]。1983年,Kabsch et al发表Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features一文,该文对氨基酸序列与蛋白质结构之间的关系进行了分析,并针对蛋白质二级结构制定了一系列简单的用于氢键键合和几何特征图像识别处理的标准[23]。1990年,Altschul et al.发表Basic local alignment search tool一文,该文提出了一种被称为基本局部比对搜索工具(BLAST)的快速序列比对方法,该工具可在DNA和蛋白质序列数据库中进行数据检索、基序检索以及基因鉴定搜索,并可对相似的长DNA序列的多个区域进行比对分析[24]。1994年,Thompson et al.发表CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice一文,作者在常用的渐进多序列比对方法基础上提出了一种蛋白质序列比对的新程序,它主要对权重的设置、氨基酸取代矩阵取向阶段的设置等作了相应的改进[25]。1995年,Murzin et al 发表SCOP: a structural classification of proteins database for the investigation of sequences and structures一文,该文提出构建一种蛋白质结构分类的数据库——SCOP,这个数据库将对已知结构的蛋白质的结构和进化关系进行详细而全面的介绍[26]。同年Benjamini et al 发表Controlling the false discovery rate: a practical and powerful approach to multiple testing一文,该文首次提出多重检验要控制伪发现率(FDR)这一概念[27],后来FDR理论与方法被广泛应用于生物海量数据统计分析中。1997年,Altschul et al.发表了经典著作Gapped BLAST and PSI-BLAST: A new generation of protein database search programs,作者提出一种运行速度是BLAST三倍的程序——PSI-BLAST,并且该程序在探测生物学相关序列相似性时更加敏感,还可以用来发现一些新的和有趣的BRCT超家族的成员[28]。以上9篇早期奠基性文献为近十年生物信息学领域的研究发展奠定了坚实的理论与方法基础,并为其指明了相应的研究方向,是近十年来生物信息学研究领域十分重要的知识基础。
3.2 高被引文献
通常,高频被引文献中传递的知识易在某一时间段内获得较多研究者的认同,并且相关研究者往往将这些高被引文献内所包含的观点、知识作为开展下一步研究的知识基础。因此,高被引文献对生物信息学领域研究具有重大的参考价值,是该领域相关研究的知识基础。利用CiteSpace软件,网络节点选择参考文献;以论文标题、摘要和关键词(包括描述词和标识符)作为前沿术语来源;将阀值设为top50;得到生物信息学研究领域文献的共被引知识图谱。

图4 国际生物信息学的文献共被引知识图谱Fig.4 Knowledge map of literatures co-citation on international bioinformatics
图4中节点的大小与节点相对应的文献被引频次成正比,节点越大表明该文献的被引频次越高。选取共被引频次不少于250的文献作为近十年国际生物信息学领域的高被引文献。通过对文献被引频次高低进行分析后发现近十年国际生物信息学领域共有15篇高被引文献。第一篇是Ashburner et al 于2000年发表的论文Gene ontology: Tool for the unification of biology,作者指出由于生物学中核心功能的基因很大一部分是由所有真核生物共享,所以知识共享这样的蛋白质在一个生物体的生物学作用往往可以转移到其他生物体[29]。第二篇是Berman et al 于2000年发表的论文The protein data bank,该文介绍了一个用来研究生物大分子的结构的数据库——PDB,文中详细介绍了PDB的建设目标,系统数据的沉积和访问以及如何获得进一步的信息的方式,除此之外还为未来资源的发展制定了近期计划[30]。第三篇是Subramanian et al 于2005年发表的论文Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles,作者在文中描述了一种解释全基因组表达谱的分析方法——基因组富集分析(GSEA),并通过实证研究证明了该方法强大的适用性[31]。第四篇是Eisen于1998年发表的论文Cluster analysis and display of genome-wide expression patterns,该文使用标准统计学算法根据基因表达谱的相似性对DNA微阵列杂交的全基因组表达数据进行了聚类分析,结果显示人类与芽殖酵母在基因表达数据组聚类上有相似的趋势[32]。第五篇是Gentleman et al.于2004年发表的论文Bioconductor: open software development for computational biology and bioinformatics,作者为计算生物学和生物信息学研发了一种开放式的软件开发工具平台——Bioconductor,该平台为计算生物学和生物信息学的可扩展的软件协同开发创造了条件[33]。限于篇幅,我们仅对前面的五篇文献做详细的说明,第6~15篇[34-43]高被引文献按共被引频次从大到小排列于表2。
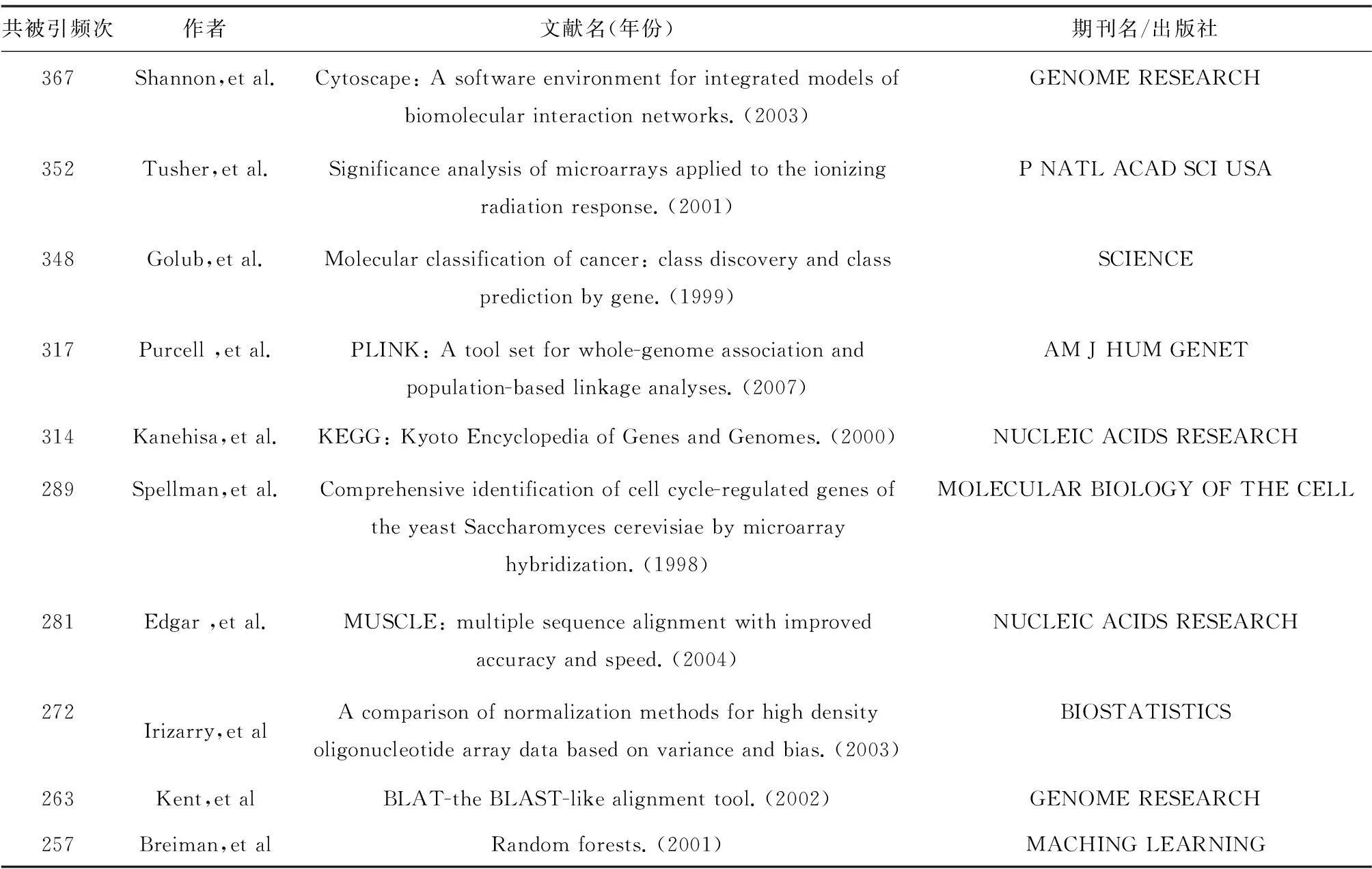
表2 国际生物信息学第6~15篇高被引文献(共被引频次≥250)
3.3 关键节点文献
文献节点中介中心性高低可反映一篇文献对学科研究领域的枢纽作用。开展生物信息学领域关键节点文献的探测,可找出一定时间内该学科领域知识演化网络中的转折点,这些转折点的节点中介中心性较高,处于不同知识聚类网络的连接路径上,可将其视为该学科交叉研究领域的重要知识基础。中心性测量为发现学科研究领域的连接关键点(演化网络中的转折点)提供了计算方法,CiteSpace将关键点的计算测量和可视属性进行合并,将中介中心性Centrality≥0.3的节点视为关键点。设置CiteSpace参数,建阀值设为top30,运行软件绘制近十年生物信息学领域关键节点文献的知识图谱,见图5。
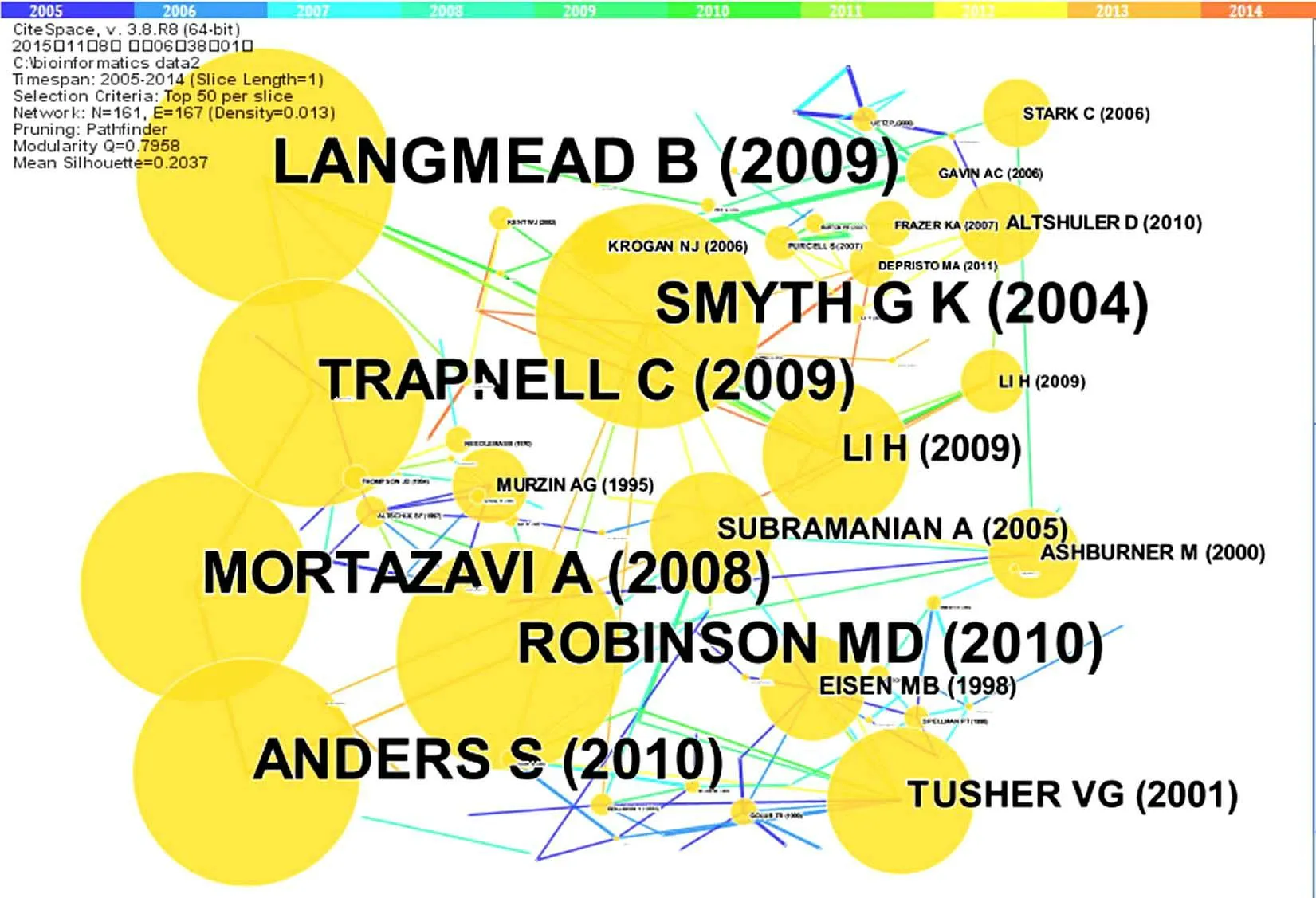
图5 国际生物信息学关键节点文献知识图谱Fig.5 Knowledge map of core literatures on international bioinformatics
分析图5中节点的中介中心性发现,近十年国际生物信息学领域关键节点(Centrality≥0.3)有8个。按照节点中介中心性大小进行排序,本文将此8篇[44-51]关键节点文献按照中介中心性大小依次排列如表3所示。

表3 国际生物信息学领域高中心度文献(Centrality≥0.3)
4结论
本文利用信息可视化计量研究方法,从多方面对国际上近十年生物信息学研究领域的研究热点、研究前沿及其知识基础进行可视化分析和展示,得到如下结论:
(1)通过绘制国际生物信息学领域的聚类视图,生物信息学研究高频词汇主要有database、identification、expression、gene-expression、protein、prediction、sequence、algorithm等,并生成5大关键词子聚类;进一步对关键词主题进行分析得出近十年国际生物信息学领域的研究热点分别是基因组与遗传学研究、蛋白组学研究、细胞与分子生物学研究、基因的数据挖掘分析、生物系统建模与仿真等。
(2)从探测研究前沿的角度出发,得出近十年国际生物信息学领域研究前沿,主要有功能基因组与比较基因组学、蛋白质结构比对与预测、分子进化分析、生物计算等领域。
(3)通过绘制近十年国际生物信息学领域文献共被引网络知识图谱,分别对被引文献进行时间、被引频次和中介中心性三方面的分析,得出最近十年生物信息学领域由9篇早期奠基性文献、15篇高被引文献和8篇高中心性关键文献构成的知识基础。
参考文献
[1]CANTOR C R,LIM H A.Electrophoresis,Supercomputing and the Hu-man genomes[M].New Jersey :World Scientific Publishing Co,1991.
[2]张春霆. 生物信息学的现状与展望 [J].世界科技研究与发展,2000,22(6):17-20.
ZHANG Chunting.The Current Status and The Prospect of Bioinformatics[J].World Sci-tech Research & Development,2000,22(6):17-20.
[3]OUZOUNIS C A,VALENCIA A. Early bioinformatics: the birth of a discipline-a personal view[J]. Bioinformatics,2003,19(17):2176-2190.
[4]PATRA S K, MISHRA S.Bibliometric study of bioinformatics literature[J].Scientometrics, 2006, 67(3): 477-489.
[5]PEREZ-IRATXETA C,ANDRADE-NAVARRO M A,WREN J D.Evolving research trends in bioinformatics[J].Briefings in Bioinformatics,2007, 8(2): 88-95.
[6]GLäNZEL W,JANSSENS F,THIJS B.A comparative analysis of publication activity and citation impact based on the core literature in bioinformatics[J].Scientometrics,2008,79(1):109-129.
[7]SONG M,KIM S Y,ZHANG G,et al.Productivity and influence in bioinformatics:A bibliometric analysis using PubMed central[J].Journal of the Association for Information Science and Technology,2014, 65(2):352-371.
[8]王玉梅, 王艳.基于文献计量的我国生物信息学研究发展动态[J].科技情报开发与经济, 2002, 12(5):1-3.
WANG Yumei,WANG Yan. Study on Developments and Tendency of Bio-information Science in Our Country Based on Literature Metrology[J].Sci-tech Information Development & Economy,2002,12(5): 1-3.
[9]宋茂海, 李东方.基于共词分析的国内生物信息学热点领域研究[J]. 生物信息学, 2014, 12(1): 46-52.
SONG Maohai, LI Dongfang. Hot spots analysis of China's bioinformatics based on co-word analysis method[J].Chinese Journal of Bioinformatics,2014,12(1): 46-52.
[10]种乐熹,胡德华. 生物信息学软件研究的可视化分析[J]. 生物信息学,2015,13(1):54-67.
ZHONG Lexi, HU Dehua. Visualizing analysis of bioinformatics software research[J].Chinese Journal of Bioinformatics,2015,13(1): 46-52.
[11]李运景,侯汉清,薛春香,等. 可视化同被引分析技术综述[J]. 图书情报工作,2008,11: 22-25.
LI Yunjing,HOU Hanqing,XUE Chunxiang,et al. Study on the Key Techniques of Co-citation Visualization[J].Library and Information Service, 2008,11: 22-25.
[12]CHEN C M.CiteSpace II:Detecting and visualizing emerging trends and transient patterns in scientific literature[J].Journal of the American Society for Information Science and Technology,2006,57(3): 359-377.
[13]赵蓉英,许丽敏. 文献计量学发展演进与研究前沿的知识图谱探析[J]. 中国图书馆学报,2010,05: 60-68.
ZHAO Rongying, XU Limin.The Knowledge Map of the Evolution and Research Frontiers of the Bibliomertrics[J]. Journal of Library Science in China,2010,05:60-68.
[14]CHEN C M.Visualising semantic spaces and author co-citation networks in digital libraries[J]. Information Processing & Management,1999,35(3):401-20.
[15]CHEN C M, PAUL R J.Visualizing a knowledge domain's intellectual structure[J].Computer, 2001, 34:65-71.
[16]邱均平,吕红. 近五年国际图书情报学研究热点、前沿及其知识基础——基于17种外文期刊知识图谱的可视化分析 [J]. 图书情报知识,2013,03: 4-15.
QIU Junping,Lü Hong.The Hot Domains,Research Fronts and Knowledge Base of International Library and Information Visua Analysis of 17 Journals’ Knowledge Map[J].Document Information & Knowledge, 2013, 03: 4-15.
[17]栾春娟,侯海燕,王贤文. 国际科技政策研究热点与前沿的可视化分析[J]. 科学学研究,2009,02: 240-243.
LUAN Chunjuan,HOU Haiyan,WANG Xianwen.Visualization Analysis of the Hot Domains and the Research Edge in the Field of S&T Policy[J].Studies in Science of Science, 2009, 02: 240-243.
[18]PERSSON O. The intellectual base and research fronts of JASIS 1986-1990 [J].Journal of the American Society for Information Science, 1994, 45(1): 31-38.
[19]赵蓉英,王菊. 图书馆学知识图谱分析[J].中国图书馆学报,2011,37(2):40-50.
ZHAO Rongying, WANG Ju. Knowledge mapping analysis of library science[J].Journal of Library Science in China, 2011, 37(2):40-50.
[20]NEELEMAN S B, WUNSCH C D. A general method applicable to the search for similarities in the amino acid sequence of two proteins[J]. Journal of molecular biology, 1970, 48(3): 443-453.
[21]DEMPSTER A P,LAIRD N M,RUBIN D B.Maximum likelihood from incomplete data via the EM algorithm[J].Journal of the Royal Statistical Society. Series B (Methodological), 1977,39(1):1-38.
[22]SMITH T F,WATERMAN M S.Identification of common molecular subsequences[J].Journal of Molecular Biology,1981,147(1):195-197.
[23]KABSCH W, SANDER C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features[J]. Biopolymers, 1983, 22(12): 2577-2637.
[24]ALTSCHUL S F,GISH W,MILLER W,MYERS E W,LIPAN D J. Basic local alignment search tool[J].Journal of Molecular Biology,1990,215(3):403-410.
[25]THOMPSON J D, HIGGINS D G, GIBSON T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice [J]. Nucleic Acids Research,1994,22(22):4673-4680.
[26]MURZIN A G,BRENNER S E,HUBBARD T.SCOP: a structural classification of proteins database for the investigation of sequences and structures[J]. Journal of Molecular Biology,1995,247(4):536-540.
[27]BENJAMINI Y, HOCHBERG Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing[J]. Journal of the Royal Statistical Society. Series B (Methodological), 1995,57(1): 289-300.
[28]ALTSCHUL S F, MADDEN T L, SCHAFFER A A, ZHANG J, ZHANG Z, MILLER W, et al. Gapped BLAST and PSI-BLAST:a new generation of protein database search programs [J]. Nucleic Acids Research,1997,25(17):3389-3402.
[29]SHBURNER M,BALL C A,BLAKE J A,BOTSTEIN D,BUTLER H,CHERRY J M,et al.Gene ontology: Tool for the unification of biology [J]. Nature Genetics, 2000, 25(1): 25-29.
[30]BERMAN H M, WESTBROOK J, FENG Z, et al.The protein data bank[J].Nucleic Acids Research,2000,28(1):235-242.
[31]SUBRAMANIAN A, TAMAYO P, MOOTHA V K, et al.Gene set enrichment analysis:a knowledge-based approach for interpreting genome-wide expression profiles[J]. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102(43): 15545-15550.
[32]EISEN M B, SPELLMAN P T, BROWN P O, et al.Cluster analysis and display of genome-wide expression patterns[J].Proceedings of the National Academy of Sciences, 1998, 95(25): 14863-14868.
[33]GENTLEMAN R C, CAREY V J, BATES D M,et al. Bioconductor:open software development for computational biology and bioinformatics [J].Genome Biology, 2004, 5(10): R80.
[34]SHANNON P, MARKIEL A, OZIER O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks[J].Genome Research, 2003, 13(11): 2498-2504.
[35]TUSHER V G,TIBSHIRANI R,CHU G.Significance analysis of microarrays applied to the ionizing radiation response[J].Proceedings of the National Academy of Sciences, 2001, 98(9): 5116-5121.
[36]GOLUB T R, SLONIM D K, TAMAYO P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring[J]. Science, 1999, 286(5439): 531-537.
[37]PURCELL S, NEALE B, TODD-BROWN K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses[J]. The American Journal of Human Genetics, 2007, 81(3): 559-575.
[38]KANEHISA M, GOTO S. KEGG: kyoto encyclopedia of genes and genomes[J]. Nucleic Acids Research, 2000, 28(1): 27-30.
[39]SPELLMAN P T, SHERLOCK G, ZHANG M Q, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization[J]. Molecular Biology of the Cell, 1998, 9(12): 3273-3297.
[40]EDGAR R C. MUSCLE: multiple sequence alignment with improved accuracy and speed[C]//Computational Systems Bioinformatics Conference, 2004. CSB 2004. Proceedings. 2004 IEEE. IEEE, 2004: 728-729..
[41]BOLSTAD B M, IRIZARRY R A, ÅSTRAND M, et al. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias[J]. Bioinformatics, 2003, 19(2): 185-193.
[42]KENT W J. BLAT—the BLAST-like alignment tool[J]. Genome Research, 2002, 12(4): 656-664.
[43]BREIMAN L. Random forests[J]. Machine Learning, 2001, 45(1): 5-32.
[44]LANGMEAD B,TRAPNELL C,POP M,et al.Ultrafast and memory-efficient alignment of short DNA sequences to the human genome[J]. Genome Biology, 2009, 10(3): R25.
[45]MORTAZAVI A, WILLIAMS B A, MCCUE K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq[J]. Nature Methods, 2008, 5(7): 621-628.
[46]TRAPNELL C,PACHTER L,SALZBERG S L.TopHat: discovering splice junctions with RNA-Seq[J].Bioinformatics, 2009, 25(9): 1105-1111.
[47]ANDERS S,HUBER W.Differential expression analysis for sequence count data[J]. Genome Biology, 2010, 11(10): R106.
[48]ROBINSON M D, MCCARTHY D J, SMYTH G K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data[J]. Bioinformatics, 2010, 26(1): 139-140.
[49]WETTENHALL J M, SMYTH G K. limmaGUI: a graphical user interface for linear modeling of microarray data[J]. Bioinformatics, 2004, 20(18): 3705-3706.
[50]TUSHER V G,TIBSHIRANI R,CHU G.Significance analysis of microarrays applied to the ionizing radiation response[J].Proceedings of the National Academy of Sciences, 2001, 98(9): 5116-5121.
[51]LI H,HANDSAKER B,WYSOKER A, et al.The sequence alignment/map format and SAMtools[J].Bioinformatics,2009,25(16):2078-2079.
Visualizing analysis of international bioinformatics research
YOU Ge,LI Yanhui*,LIU Xiang
(SchoolofInformationManagement,CentralChinaNormalUniversity,Wuhan430079,China)
Abstract:The current well-known information visualization software CiteSpace was used for statistical analysis and visualization for papers published in 5 high-impact international SCI journals from 2005 to 2014 in the field of bioinformatics, draw the network visualization patterns of keyword co-occurrence, bursting word co-occurrence,classic literature co-occurrence,highly cited literature co-occurrence and core literatures co-occurrence to reveal hot research topics and knowledge base of international bioinformatics for helping researchers to understand the trend of the research.
Keywords:Bioinformatics;CiteSpace;Information visualization;Knowledge mapping;Hot topics
中图分类号:G350
文献标志码:A
文章编号:1672-5565(2015)04-257-09
doi:10.3969/j.issn.1672-5565.2015.04.09
作者简介:游鸽,男,硕士研究生,研究方向:生物信息学与数据挖掘;E-mail:374005361@qq.com.*通信作者:李延晖,男,教授,博士生导师,研究方向:生物仿真与数据挖掘;E-mail:yhlee@mail.ccnu.edu.cn.
收稿日期:2015-07-30;修回日期:2015-11-13.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
师道·教研(2022年1期)2022-03-12
现代畜牧科技(2021年4期)2021-07-21
少先队活动(2020年12期)2021-01-14
海洋信息技术与应用(2020年1期)2020-06-11
传媒评论(2019年4期)2019-07-13
中国博物馆(2018年2期)2018-12-05
中成药(2017年3期)2017-05-17
领导科学论坛(2016年9期)2016-06-05
现代检验医学杂志(2015年4期)2015-02-06
