
反向虚拟筛选平台及应用
2015-02-23 07:58刘雨桐宋新蕊周雪松
生物信息学 2015年4期
关键词:毒副作用
张 媛,刘雨桐,李 达,宋新蕊,周雪松,赵 勇
(北京市计算中心,北京 100094)
反向虚拟筛选平台及应用
张媛*,刘雨桐*,李达,宋新蕊,周雪松,赵勇**
(北京市计算中心,北京 100094)
摘要:靶标确证是老药新用、药物毒副作用研究的关键。基于分子对接方法AutoDock Vina和内部构建的疾病靶标数据库,采用分布式架构,构建了反向虚拟筛选平台。应用该平台对药物吡斯的明进行靶标确证,最终成功找到其靶标乙酰胆碱酯酶,验证了平台的实用性和准确性。
关键词:老药新用;毒副作用;反向虚拟筛选;AutoDock Vina
随着计算机科学和生物信息学的迅猛发展,越来越多的生命科学数据被挖掘出来,常规药物发现过程面临投资大和回报周期长的问题,计算机模拟药物设计通过辅助药物发现的靶标识别与确证、先导化合物发现与优化、临床前研究等模块,大大节省了药物发现的时间与资金投入,成为生命科学领域最为前沿的技术之一。
近十年,药物多靶标的研究成为越来越热门的话题,截止目前,DrugBank数据库中的7 759种药物有28.5%(2 245个)是多靶标作用[1]。药物作用于多个靶标是药物毒副作用的根源,也是药物新作用研究(老药新用)的基础,对于药物研发有着重大意义。
反向虚拟筛选(Reverse virtual screening)是一种对给定药物或活性小分子通过计算方法找到其潜在药物靶标的技术[2]。对于已知药物,反向虚拟筛选技术可以寻找其他潜在靶标,为药物新作用研究、药物毒副作用研究提供参考;对于活性小分子,反向虚拟筛选技术可以预测其潜在作用靶标,为活性化合物的后期改造和机制研究提供方向,尤其适用于药理活性明确但靶标未知的中药等的研究(见图1)。
反向虚拟筛选技术越来越多地应用于药物研发的各个方面。Bernard等运用反向分子对接方法发现了托非索泮(Tofisopam)的作用靶点[3]。托非索泮是一种外消旋体,80年代作为抗焦虑药在亚洲和欧洲上市。托非索泮作用于磷酸二酯酶4,其S 型异构体与已上市的PDE4抑制剂咯利普兰相比,抑制作用更强,且不存在呕吐的副作用,为PDE4的高选择性抑制剂的研究提供了参考意义。张明波等人对中药中的一种活性物质槲皮素进行了研究[4],他们利用反向对接的方法把槲皮素与抗肿瘤靶标进行反向对接,确定了槲皮素抗肿瘤可能的作用靶标和分子机制,为槲皮素的抗肿瘤药物研发奠定了基础。林爱斌等人利用反向对接的方法预测异白花丹酮的人类疾病靶点[5],发现Vitamin D3 receptor GTPase Hras 和 Leuko-cyte elastase 可能是异白花丹酮的人类疾病靶点。许先进等人对持久性有机污染物4,4’-DDE和CB-153利用反向虚拟筛选的方法搜索生物体内潜在蛋白质受体[6],发现反向虚拟筛选的方法在理解污染物分子的致病机制方面有很大的帮助。Gianluigi Lauro等人对天然产物黄腐酚和异黄腐酚针对肿瘤相关靶标进行了反向虚拟筛选[7],发现了黄腐酚和异黄腐酚具有激酶活性,进一步证实了反向虚拟筛选方法的鲁棒性。
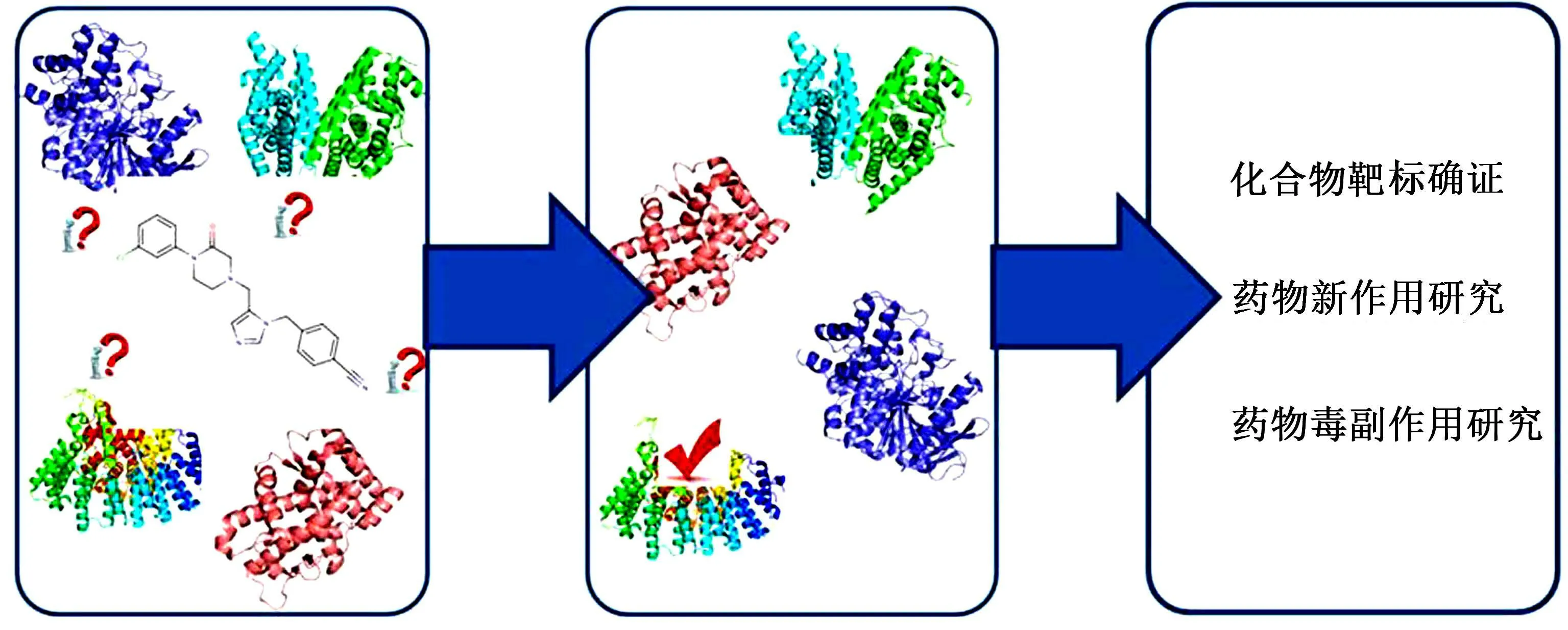
图 1 反向虚拟筛选和用途Fig.1 Reverse Virtual Screening and its usage
反向虚拟筛选分为基于受体的反向虚拟筛选和基于配体的反向虚拟筛选两类。基于受体的反向虚拟筛选是将化合物一一对接到靶标的活性位点处,根据化合物-靶标的相互作用能或者药效团匹配得分排序,得分越高,潜在靶标的可能性越大。常见的基于受体的反向虚拟筛选方法有idTarget[8],TarFisDock[9],Mdock[10]和PharmMapper[11]。
基于配体的反向虚拟筛选基于“Chemical similarity principle”原理[2],认为相似的化合物具有相似的化学性质,甚至有着相似的作用靶标。因此将提问化合物与数据库中的化合物一一进行相似性匹配打分,相似性最高的化合物作用靶标即认为是提问化合物的潜在作用靶标。ChemMapper是基于化合物形状和药效匹配的反向虚拟筛选方法[12]。
反向虚拟筛选方法的成功率一方面取决于筛选方法的准确性,另一方面取决于数据库构建的完整性和准确性。IdTarget采用改良的AutoDock方法,数据库包含PDB库中所有晶体结构,靶标针对性较差;TarFisDock基于DOCK方法[13],使用PDTD数据库[14],该数据库涵盖了841个已知或者潜在药物靶标及其治疗领域,针对性完整性较好。
为了保证靶标数据库的针对性和准确性,同时考虑靶标的结构完整性,我们选择现有人类基因组的可成药基因在PDB数据库中有完整解析和部分解析的蛋白质结构构建了疾病靶标数据库,同时,我们应用AutoDock Vina作为分子对接工具[15],结合疾病靶标数据库构建了反向虚拟筛选平台(http://reversedock.vslead.com/)。应用该平台对药物吡斯的明进行靶标确证,成功找到其靶标乙酰胆碱酯酶,验证该平台的实用性和准确性。
1材料与方法
反向虚拟筛选平台关键数据在于疾病靶标数据库的建立,该数据库包含pdbqt格式的靶标三维晶体结构、活性位点参数文件和靶标生物信息,分别从靶标三维晶体结构获取、靶标活性位点确定和靶标生物信息收集三方面来进行(见图2)。
1.1 数据来源
DGIdb是一个鉴定已知的和潜在的药物-基因关系的网络数据库[16],其中收集的药物-基因相互作用数据有多个来源,包括DrugBank[1]、TTD[17]、PharmGKB[18]等。从DGIdb数据库中搜集人类基因组的可成药基因,共获取6 571条基因,筛选数据质量可靠的基因条目,通过NCBI网站获取对应的蛋白序列,共得到12 946条蛋白序列。
1.2 获取靶标三维晶体结构
应用本地Blast方法,以12 946条蛋白序列为查询序列,从PDB数据库中搜索相似序列,对于每一个匹配结果进行分析过滤。为了获取全部基因序列和部分连续基因序列被解析出三维结构的靶标,避免由于三维结构的不连续而导致的筛选结果不可靠,选择PDB数据库中完全匹配的蛋白作为有完整解析结构的蛋白,将匹配长度小于查询序列,但匹配度为100%并且gap是0,错配是0的蛋白作为有部分解析结构的蛋白。

图2 反向虚拟筛选平台构建方法Fig.2 Method of Reverse Virtual Screening platform
1.3 确定靶标活性位点
获取疾病靶标的三维结构后,对靶标的活性位点进行确定。活性位点确定的方法主要有两种,一种方法是以小分子周围范围残基为活性位点残基,此方法找到的活性位点多是生物活性位点,缺点是该靶标必须有蛋白—小分子复合物晶体结构。另一种是通过活性位点预测软件预测得到,如LIGSITE[19]、PocketPicker[20],该方法适用于所有有晶体结构的靶标,缺点是易出现假阳性活性位点。为确保口袋的准确性,采取第一种方法提取小分子6埃范围内残基为活性位点残基,以其所占立方体划定盒子,确定筛选活性位点中心和尺寸。
1.4 收集靶标生物信息
从PubMed、UniProt、PDB等数据库收集靶标的对应数据库编号,通过数据库检索,可得到靶标的文献、序列、结构等信息;另外,从DrugBank数据库收集了靶标对应药物的CAS号、DrugBank编号、药物名称信息,为用户更全面了解疾病靶标性质提供参考。
1.5 反向虚拟筛选平台的构建
在内部构建数据库的基础上,应用AutoDock Vina方法,基于分布式虚拟筛选架构,结合高性能计算集群优势,构建了反向虚拟筛选平台。用户提交小分子化合物文件、选择要筛选的内部靶标数据库(Whole或者part)、确定返回结果数(最多前100结果)即可提交反向虚拟筛选任务(见图3)。
2结果与分析
2.1 疾病靶标数据库构建结果
构建了600多个靶标7 000多个活性位点的疾病靶标数据库,其中具有完整结构的靶标有140个,部分结构的靶标有506个(见表1)。此外,还给出每个靶标的对应PubMed、EC、UniProt、CAS、DrugBank等生物信息学数据,为医药工作者的药物研发提供帮助。

表 1 疾病靶标数据库
2.2 反向虚拟筛选平台构建结果
成功构建了反向虚拟筛选服务平台。用户只需提供小分子化合物结构,即可得到其潜在作用靶标(见图4)。
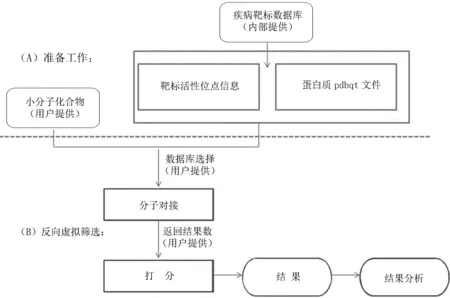
图3 反向虚拟筛选平台工作流程Fig.3 Reverse virtual screening platform workflow

图4 反向虚拟筛选平台页面Fig.4 Reverse virtual screening interface
3反向虚拟筛选平台应用
药物吡斯的明对眼有缩瞳、降低眼内压的作用,作为抗胆碱酯酶药使用,其作用靶标是乙酰胆碱酯酶。用吡斯的明作为提问化合物,应用反向虚拟筛选平台进行筛选,验证该平台是否能找到吡斯的明的作用靶标乙酰胆碱酯酶。
从DrugBank数据库下载得到吡斯的明的sdf格式结构(见图5a),针对part靶标数据库,应用反向虚拟筛选平台提交筛选任务,最后选择返回前100结果。
结合模式图(见图5b)可以看到,吡斯的明的酯键与Ser125和Tyr337形成H键相互作用,苯环与Trp86形成好的pi-pi相互作用。
结果显示,吡斯的明成功找到了其作用靶标乙酰胆碱酯酶。此外,结果文件还给出了乙酰胆碱酯酶的PubMed数据库、UniProt数据库、DrugBank数据库的相关信息,为靶标分析提供更多参考(见图6)。

图5 吡斯的明结构和其结合模式图Fig.5 Pyridostigmine structure and binding mode
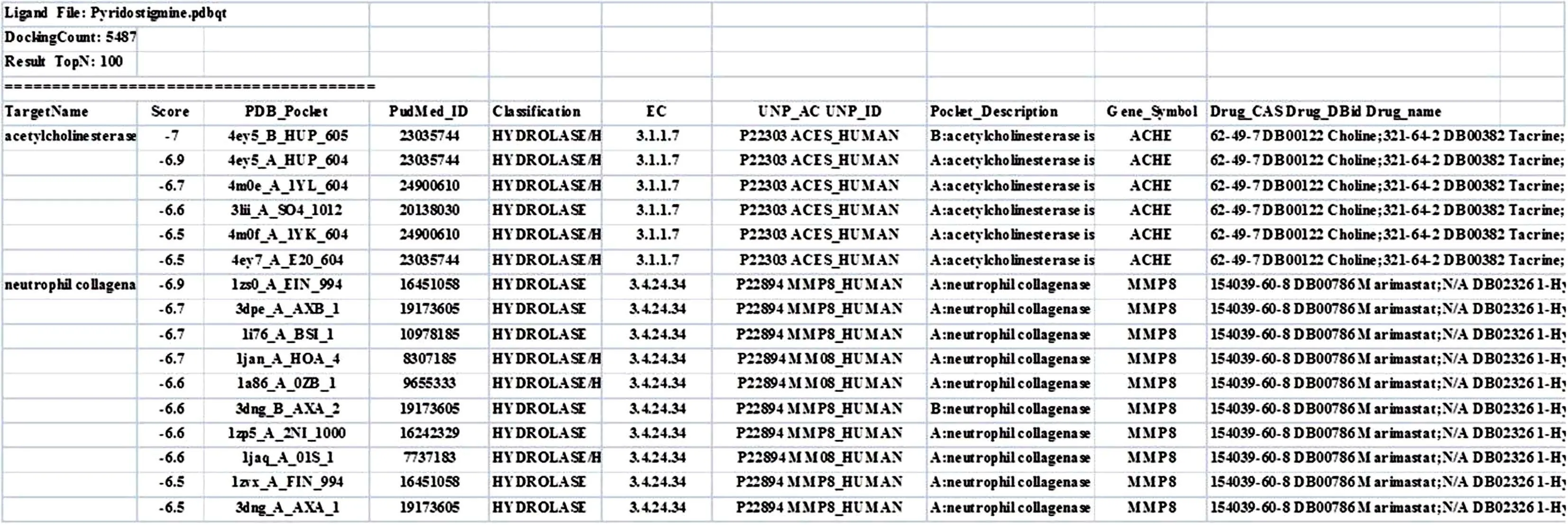
图6 吡斯的明反向虚拟筛选结果示例Fig.6 Result example of pyridostigmine reverse virtual screening
4结论
成功构建了疾病靶标数据库,该数据库包含了600多个疾病靶标的7 000多个活性位点,为化合物的疾病治疗提供参考;成功构建了反向虚拟筛选平台,应用该平台成功找到了药物吡斯的明的靶标乙酰胆碱酯酶,为反向虚拟筛选平台的应用提供基础。该平台具有良好的实用性和准确性。
参考文献
[1]LAW V, KNOX C, DJOUMBOU Y, et al. DrugBank 4.0: shedding new light on drug metabolism[J].Nucleic Acids Research, 2014, 42: 1091-1097.
[2]CERETO-MASSAGUE A, OJEDA M J, VALLS C, et al. Tools for in silico target fishing[J].Methods, 2015, 71: 98-103.
[3]BERNARD P,DUFRESNE-FAVETTA C,FAVETTA P,et al. Application of drug repositioning strategy to TOFISOPAM[J].Current Medicinal Chemistry, 2008, 15(30): 3196-3203.
[4]张明波 李莉, 孙艳涛, 等. 基于分子反向对接方法的槲皮素抗肿瘤机制研究[J].中国实验方剂学杂志, 2012, 23: 145-148.
ZHANG Mingbo, LI Li, SUN Yantao, et al. Study on Antitumor Mechanism of Quercetin based on Molecular Reverse Docking Method[J]. Chinese Journal of Experimental Traditional Medical Formulae, 2012, 23: 145-148.
[5] 林爱斌 刘亮, 姚晓敏, 等. 异白花丹酮的人类疾病靶点预测研究[J].中药药理与临床, 2015, 01:63-66.
LIN Aibing, LIU Liang, YAO Xiaomin, et al. Study on human disease-related target prediction of isozeylanone[J].Pharmacology and Clinics of Chinese Materia Medica,2015,01: 63-66.
[6] 许先进 苏计国, 刘斌, 等. 持久性有机污染物4,4'-DDE和CB-153的反向虚拟筛选[J].物理化学学报, 2013, 10:2276-2285.
XU Xianjin,SU Jiguo,LIU Bin,et al.Reverse virtual screening of persistent organic pollutants 4,4'-DDE and CB-153[J].Acta PhysicoChimica Sinica,2013,10:2276-2285.
[7]LAURO G,MASULLO M,PIACENTE S,et al. Inverse Virtual Screening allows the discovery of the biological activity of natural compounds[J].Bioorganic & Medicinal Chemistry,2012,20(11): 3596-3602.
[8]WANG J C,CHU P Y,CHEN C M, et al. idTarget:a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach[J].Nucleic Acids Research,2012,40:393-399.
[9]LI H, GAO Z,KANG L,et al.TarFisDock:a web server for identifying drug targets with docking approach[J].Nucleic Acids Research, 2006, 34: 219-224.
[10]HUANG S Y and ZOU X.Construction and test of ligand decoy sets using MDock: community structure-activity resource benchmarks for binding mode prediction[J].Journal of Chemical Information and Modelling,2011,51(9):2107-2114.
[11]LIU X, OUYANG S,YU B,et al. PharmMapper server:a web server for potential drug target identification using pharmacophore mapping approach[J].Nucleic Acids Research,2010,38:609-614.
[12]GONG J,CAI C,LIU X,et al.ChemMapper:a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method[J].Bioinformatics,2013,29(14):1827-1829.
[13]EWING T J, MAKINO S, SKILLMAN A G, et al.DOCK 4.0:search strategies for automated molecular docking of flexible molecule databases[J].Journal Computer Aided Molecular Design,2001,15(5):411-428.
[14]GAO Z, LI H, ZHANG H, et al. PDTD: a web-accessible protein database for drug target identification[J].BMC Bioinformatics,2008,9:104.
[15]TROTT O and OLSON A J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading[J].Journal of Computational Chemistry, 2010, 31(2): 455-461.
[16]GRIFFITH M, GRIFFITH O L, COFFMAN A C, et al. DGIdb: mining the druggable genome[J].Nature Methods, 2013,10(12):1209-1210.
[17]QIN C,ZHANG C,ZHU F,et al.Therapeutic target database update 2014:a resource for targeted therapeutics[J].Nucleic Acids Research, 2014, 42:1118-1123.
[18]THORN C F, KLEIN T E and ALTMAN R B.PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base[J].Methods in Molecular Biology, 2005, 311:179-191.
[19]HENDLICH M, RIPPMANN F,BARNICKEL G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins[J].Journal of Molecular Graphics and Modelling, 1997, 15(6): 359-363, 389.
[20]WEISEL M, PROSCHAK E and SCHNEIDER G. PocketPicker: analysis of ligand binding-sites with shape descriptors[J].Chemistry Central Journal, 2007,1: 7.
更正:本刊2007年第5卷第4期P157:“1.2置jdk1.4.2”更正为“1.2配置jdk1.4.2”。
Reverse virtual screening platform and its application
ZHANG Yuan*, LIU Yutong*,LI Da, SONG Xinrui, ZHOU Xuesong, ZHAO Yong**
(BeijingComputingCenter,Beijing100094,China)
Abstract:Computer based-target identification provides a unique tool for finding new application of old drugs and for the research on side effects of drug toxicity. Using molecular docking software AutoDock Vina and an in-house disease target database, we developed a Reverse Virtual Screening platform to identify protein targets for small molecules.The platform has been tested with pyridostigmine,a drug targeting acetylcholinesterase. This Reverse Virtual Screening successfully identified acetychlinesterase as pyridostigmine’s target,showing its applicability and accuracy in computer assisted drug design.
Keywords:New application of old drug;Side effects; Reverse virtual screening;AutoDock vina
中图分类号:R91
文献标志码:A
文章编号:1672-5565(2015)04-251-06
doi:10.3969/j.issn.1672-5565.2015.04.08
作者简介:张媛,女,硕士研究生,计算化学工程师,研究方向:反向虚拟筛选;E-mail:zhangyuan@bcc.ac.cn.*同等贡献作者:刘雨桐,女,本科生,研究方向:生物信息;E-mail:liuyutong1008@hotmail.com.**通信作者:赵勇,男,博士,北京市特聘专家,研究方向:生物信息学与计算药物;E-mail:zhaoyong@bcc.ac.cn.
基金项目:北京市东城区科技计划(2015-2-006)支持;北京市科学技术研究院创新团队计划(IG201305C2)支持。
收稿日期:2015-09-29;修回日期:2015-11-17.
猜你喜欢
现代畜牧科技(2021年7期)2021-07-28
中外医学研究(2017年36期)2018-01-26
中国医药导报(2017年23期)2017-09-22
中西医结合心血管病电子杂志(2016年29期)2017-06-20
中国民族民间医药·上半月(2016年12期)2017-01-11
中国实用医药(2016年26期)2016-11-07
中国卫生标准管理(2015年18期)2016-01-20
中国医药科学(2015年14期)2015-09-15
爱你(2015年5期)2015-07-05
爱你(2015年10期)2015-04-19
