
蛋白质折叠识别方法综述
2015-02-23 07:58鄢仁祥王晓峰许伟明蔡伟文
生物信息学 2015年4期
鄢仁祥,王晓峰,许伟明,林 娟,蔡伟文
(1.福州大学生物科学与工程学院,福州 350108;
2.山西师范大学计算机与数学学院,山西 临汾 041004)
蛋白质折叠识别方法综述
鄢仁祥1,王晓峰2,许伟明1,林娟1,蔡伟文1
(1.福州大学生物科学与工程学院,福州 350108;
2.山西师范大学计算机与数学学院,山西 临汾 041004)
摘要:蛋白质折叠识别算法是蛋白质三维结构预测的重要方法之一,该方法在生物科学的许多方面得到卓有成效的应用。在过去的十年中,我们见证了一系列基于不同计算方式的蛋白质折叠识别方法。在这些计算方法中,机器学习和序列谱-序列谱比对是两种在蛋白质折叠中应用较为广泛和有效的方法。除了计算方法的进展外,不断增大的蛋白质结构数据库也是蛋白质折叠识别的预测精度不断提高的一个重要因素。在这篇文章中,我们将简要地回顾蛋白质折叠中的先进算法。另外,我们也将讨论一些可能可以应用于改进蛋白质折叠算法的策略。
关键词:折叠识别;序列比对;结构预测
1 INTRODUCTION
Protein three-dimensional(3D)structures contain essential information to characterize the protein functions. The 3D structure of a protein can be obtained through wet experimental methods,including X-ray crystallography[1]and NMR spectroscopy[2].Unfortunately,wet experiments for protein 3D determination are generally time-consuming,and,moreover,such experiments usually require amazing funding, especially for some membrane proteins,such as G protein-coupled receptors(GPCRs)[3].Alternately,protein 3D structures can also be predicted by computational algorithms, which aim to predict the correct 3D structure of a protein from its primary sequence.In fact,protein structure prediction methods have been widely used and show effective performance in many aspects of biological sciences[4,5].
Depending on utilizing algorithms,protein structure prediction methods can be roughly divided into three categories:(1)homology modeling,(2)fold recognition,(3)free modeling.Both homology modeling and fold recognition can be regarded as template-based modeling methods[6,7].The most difference between homology modeling and fold recognition is that structural- and profile-based terms are intensively used in fold recognition while homology modeling mainly relied on the information of protein sequences. Homology modeling methods are designed for targets that can find closely homologous templates, and such query proteins are usually called ‘Easy’ targets. However, it is possible that two structurally similar proteins may share weak sequence similarity (i.e., remote homologs). In the context, fold recognition methods are designed to identify remotely homologous templates. Unlike template-based methods, free modeling, which is also calledabinitioprotein folding, can be built from scratch and closely homologous templates are not required in the meantime.However,free modeling generally requires longer computational time and more computational resources, which restricts its success only to small proteins.Free modeling algorithms are only successfully applied to small proteins because of such restrictions.
As clearly pointed out by Baker and Sali[8],template-based modeling is the most reliable approach to protein 3D structure prediction.Moreover,in our previous work,we have proven that the qualities of protein 3D models by template-based modeling methods can be significantly increased when sequence alignments are as accurate as structure alignments[9].Compared to homology modeling, fold recognition is more challenging, exciting and important in the current post-genomic era. Profile-based methods are widely used in fold recognition. Profiles are usually calculated from multiple sequence alignments(MSAs)obtainedby PSI-BLAST[10].Two types of sequence profiles exist. One is position-specific scoring matrix(PSSM), the other is hidden Markov model(HMM). HMMs can be represented by a chain of match and insert/deletion nodes with the MSAs[9].
During the last decade, a variety of fold recognition methods have been elegantly developed(e.g., FFAS[11],FFAS-3D[12],HHsearch[13],RaptorX[14],DescFold[15],MUSTER[16]and SPARK-X[17]) and some elegant web servers have also been freely accessible to the research community. FFAS probably is the first publicly available profile-profile alignment web server for fold recognition.FFAS only uses sequence profile in its scoring function. Xuetal. extended the algorithm and proposed a new algorithm called FFAS-3D by including structural terms for identifying remote homologs.In addition,Zhou group developed a series of profile-profile alignment methods for fold recognition,including SPARK,SPARK2,SPARK3,SPARK5 and SPARK-X[18].The SPARK-X is the newest version of the SPARK series programs.HHsearch is an HMM-HMM alignment method. There are some differences among these methods. In the following sections of this manuscript,we will review representatives of these classical and popular fold recognition methods.
2Representative protein fold recognition methods
2.1 A general flow chart for the development of fold recognition methods
Although different computational techniques were used by different research groups to develop effective fold recognition algorithms,these methods usually can be summed up in a similar procedure(Fig.1).
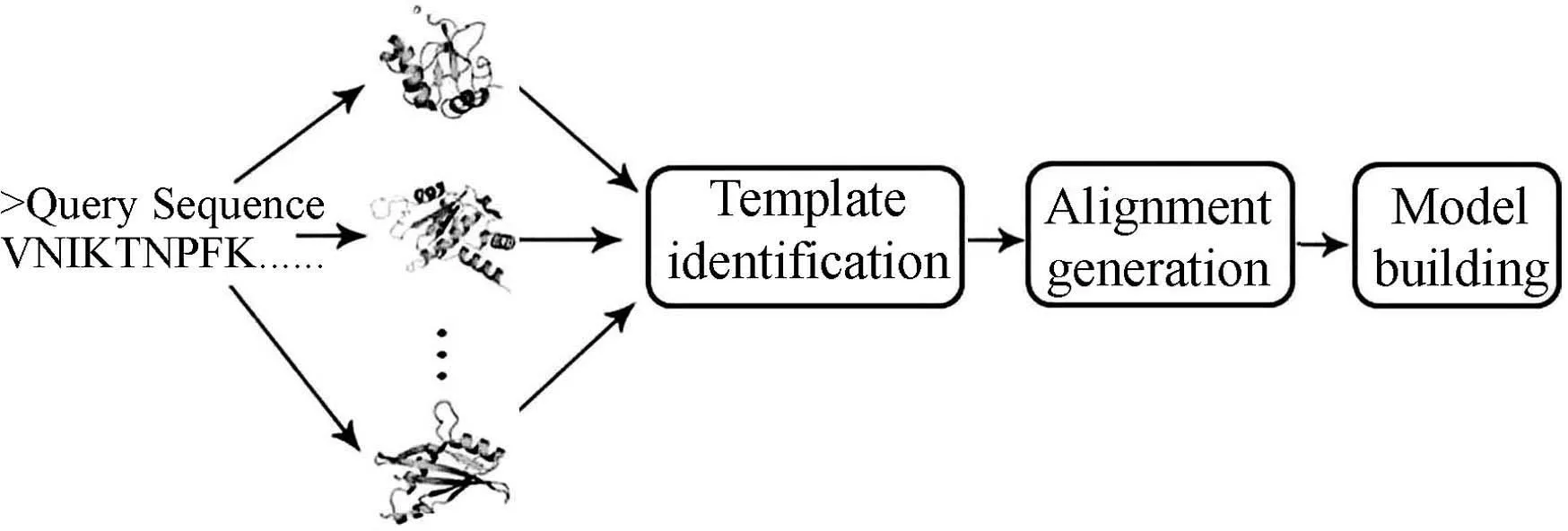
Fig.1 A general flow chart for protein foldrecognition methods
First,a(remotely) homologous protein with known structure is identified as a template from Protein Data Bank(http://www.rcsb.org/pdb/home/home.do) for a query sequence based on sequence similarity and sequence-structure compatibility. To make the template identification fast and reliable,a filtered database of Protein Data Bank with sequence identity less than 70% or 50% is usually employed.Meanwhile,almost all the major fold recognition programs used scores derived from profile-profile alignment to identify templates.Machine learning algorithms, such as neural network[19],random forest[20]and support vector machine[21]have also been employed to develop scoring functions for template identification. The scoring function developed here can be regarded as a measure to evaluate how well the query sequence fits into a structurally known protein.The measures to select suitable templates usually rely on match ZScore or e-value (Fig.2).

Fig.2 Alignment measures to select templates
The templates with ZScore or e-value better than pre-defined cutoffs are selected.The second step is to obtain an optimal alignment between the query and the template sequences. The accuracy of the alignment is highly important to the model building. Finally, several full-length and refined 3D models of the query protein are built based on the satisfaction of spatial restraints[22]by considering template atom coordinates and the alignment of query-template. In Fig.1, we summarize this general flowchart for the development of typical fold recognition methods. Furthermore, it is valuable for readers to know the publicly available and state-of-the-art web servers/tools for fold recognition methods. Here, several tools and their web servers are listed in Table 1.
Representatives of these popular fold recognition methods are introduced as follows.
2.1.1FFAS and FFAS-3D
FFAS[11]is a simple profile-profile alignment program without using structural information.First,FFAS obtains multiple sequence alignments(MSAs) by PSI-BLAST searching against a database called NR85s database with 5 iterations with an e-value threshold of 0.001.Second,FFAS uses a similar way to Henikoff weight to calculate sequence profiles.Then, a dot-product scoring function is used to align two sequence profiles.The scoring functionS(i,j) for aligning theith residue on the query and thejth residue on the template of FFAS is as

Table 1 State-of-the-art fold recognition methods
(1)
whereffq(i,k) andfft(j,m) stand for the sequence frequency ofkth residue at theith position andmth residue at thejth position for query and template profiles, respectively.Finally, the significance of alignment scores is calculated by comparing the protein with the distribution of scores obtained from pairs of unrelated proteins. FFAS is widely used by many labs in many applications. Recently, Xu et al. extended the FFAS method and developed a method called FFAS-3D by using structural features. It is reported that the performance of FFAS-3D is much better than that of FFAS, especially for hard targets. The scoring function of FFAS-3D is as
ffq(i,k)BL(k,m)fft(j,m)

(2)
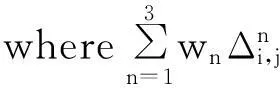
2.1.2SPARK-X
SPARK-X is a proven fold recognition method by using probabilistic-based alignment between predicted properties of query and corresponding native properties of templates. There are several variants of SPARKS developed by Zhou group, such as SPARK2, SPARK3 and SPARK5. Among them, SPARK-X is the newest version. The performance of SPARK-X mainly relies on the significantly improved predictions of structural properties, such as secondary structure, real value torsion angle and solvent accessibility.Similar to FFAS-3D, SPARK-X is also a structurally enhanced profile-profile alignment method. The scoring function used in SPARK-X is as
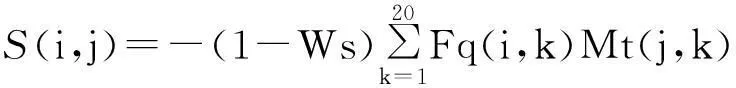
+shift
whereFq(i,k) represents the frequency of thekth amino acid at theith position of the MSAs obtained by PSI-BLAST search for a query sequence against the NCBI[23]NR database for 3 repeats with an E-value threshold of 0.001.Mt(j,k) represents the value in the log-odd profile of the template for thekth amino acid at thejth position. Similarly,Ft(j,k) represents the frequency of thekth amino acid at thejth position for the template.Mq(i,k) represents the log-odd profile of the template for thekth amino acid at theith position.Ws,w1,w2andw3are weights to sum up different terms.The shift is used to avoid aligning any unrelated pairs.
2.1.3FUGUE
FUGUE[6]is a fold recognition method that can search sequences against fold libraries by utilizing environment-specific substitution tables and structure-dependent gap penalties, where amino acid matching scores and insertions/deletions penalties are evaluated depending on the local environment of each amino acid residue in a known structure[6].Fold library and substitution tables used in FUGUE are derived from the HOMSTRAD[24]database. Meanwhile, FUGUE can automatically select alignment algorithms with detailed structure-dependent gap penalties. FUGUE has been used as one module in the Sybyl-X (http://www.certara.com/products/molmod/sybyl-x) commercial package.
2.1.4GenTHREADER
GenTHREADER[25]is developed by employing a simple neural network[19]to combine various sources of information, such as sequence alignment score, sequence length and energy potentials derived from threading, to a final score, which represents the homologous relationship between two proteins.The input features are directly fed into the input layer.The standard sigmoid activation function is used in the GenTHREADER. There are two nodes in the output layers.In the training process,the two nodes of output layers are encoded as(1,0) for structurally related protein pairs,and (0,1) for structurally unrelated protein pairs.The advantage of GenTHREADER is that various types of information can be used, including profile-profile alignment and other global features of proteins.
2.1.5Raptor
Raptor[14,26,27]is a novel method based on the mathematical theory of linear programming approach to predict 3D models of proteins via fold recognition. In Raptor method, the protein fold recognition problem is solved by a large scale integer programming problem. The profile-profile alignment,secondary structure and contact map terms, etc, are used in Raptor. RaptorX[14],which is one variant of Raptor method, excels at predicting ‘hard’ targets according to the 2010 CASP9[28]experiments(Table 2).

Table 2 Scoring terms used in single fold recognition methods
2.1.6Meta methods
Diverse methods are widely used in fold recognition algorithms and they clearly demonstrated their amazing success in the CASP[29]and CAFASP[30]competitions as well as in real-time LiveBench experiments[15,31].Some alignment-free template selection methods have also been proposed, such as motif-based fold assignment[32]and pattern-based protein folds similarity identification[33].Alignment-dependent and alignment-free methods may be complementary. Meta methods, which sometimes are also called consensus methods, are developed by combining well-established and complementary programs, aiming to further improve the accuracy of 3D model building by taking into account the outputs of existing methods. It is found that meta or consensus methods usually perform better than any single method that consists of them. The main idea of meta methods is intensively exploiting the complementarity of different methods to enhance the accuracy of template identification and query-template alignment generation. For example, LOMETS[34](Local Meta-Threading-Server) obtains 3D models by using alignments from locally-installed fold recognition programs, including FFAS-3D[12],HHsearch[13],MUSTER[16],pGenTHREADER[35],PPAS[36],PRC[37],PROSPECT[38],SP3[18], and SPARKS-X[17].The web server of LOMETS is publicly available at http://zhanglab.ccmb.med.umich.edu/LOMETS/.Pcons[39]is also a meta algorithm consisting of multiple methods. Pcons selects the best 3D models out of those produced by six prediction servers by using a neural network[40].Developers are usually required to carefully tune the parameters of meta methods to obtain the optimal performance.
2.2 Development of new fold recognition methods
By analyzing existing methods, the following aspects probably can be considered to develop improved fold recognition method.
2.2.1Novel neural network based scoring function
Most dynamic programming alignment methods[41]propose scoring functions to simply add different terms as
score(i,j) = a + b + c + shift
(4)
wherea,bandcrepresent profile, secondary structure- and other structural property-based terms,respectively.The shift is a constant value to avoid aligning unrelated residues.Theoretically, such combination may be not globally optimal. To combine different measures to make a final decision, neural network may be one of the best choices. Therefore, a neural network-based scoring function probably can be used through the following procedure. First, compile a large number of non-redundant structurally known proteins and structurally align them. Second, select the homologous protein pairs and extract the structure-based sequence alignments. If the distances of aligned residue pairs are less than a cutoff (e.g., 5.0 Å),we can consider them ‘positive’ residue pairs, otherwise, ‘negative’ pairs. Then, those terms of the scoring function can be normalized to the range of 0-1 and fed into the neural network to train the novel scoring function for any two residues. To make this idea more clear, we draw Fig.3 to demonstrate it. Although the neural network algorithm has been employed in the template identification, to the best of our knowledge, the algorithm has not been in the scoring function of profile-profile alignments.
2.2.2Respective models for template selection and sequence alignment
In a fold recognition-based protein structure prediction procedure,correctly recognizing a suitable template for a query sequence is the first step.The second step is to obtain optimal sequence alignment between the query and the template sequences.Both template identification and alignment generation are crucial to the final quality of 3D models.In most methods,however,the trained parameters obtained in sequence alignment are directly used in the template selection.A potential problem existing here is that the parameters trained for alignment generation probably are not the most optimal for template selection.Both sets of parameters probably can be optimized independently,which may obtain better performance.To get a high quality model,template selection and alignment generation are nearly equally important.New fold recognition algorithms designed with independent scoring functions for template selection and alignment generation may obtain higher accuracy.
Two nodes in the output layer,R and U representing related and unrelated residue pairs,respectively.Different features, such as sequence profile-,secondary structure-,contact map- and structural feature-based terms can be fed into the neural network to train the neural network model.Finally,the values output by the trained neural network can be used as a scoring function in the profile-profile alignment.

Fig.3 A novel neural network-based scoring function
2.2.3Dynamic selection of scoring functions and gap penalties
Dynamic scoring functions can be employed for different cases.For example,if query and template are very similar at the sequence level,the predicted structural terms can be removed according to the fact that the prediction accuracy of structural information for query sequence has not reached a satisfactory performance and they may introduce noise in such cases.Probably,the following in-depth procedure can be used.When the global sequence alignment identity of query-template is higher than 30%,sequence-sequence alignment method can be directly used to build the 3D models.When identity between sequence and structure is less than a pre-defined threshold and profiles alignment scores are significant,a sequence based profile-profile alignment scoring function such as that of FFAS can be employed.Profile-profile alignment with structural information terms considered should be used as a last option.Generally,the employed gap penalty model is affine gap penalty as
penalty = g + r(x-1)
(5)
wheregis gap opening penalty andris gap extension penalty with the constrain of |g|>|r|.xis the length of gap. Meanwhile, position-dependent gap penalty is also used by some methods. For example,Shietal. proposed a structure-dependent gap penalty method[6], in which the gap penalty for each position is dynamically changed according to its solvent accessibility, its position relative to the secondary structure elements (SSEs) and the conservation of the SSEs. Some methods employed gap penalty models derived from evolutionary or secondary structure information. Zhou group developed a position-specific gap penalty model[42], in which gap scoring scheme is derived from statistical analysis of gaps in the MSAs created by PSI-BLAST.Zhang group presented a similar structural position-dependent gap penalty method, in which no gap is allowed inside the secondary structure regions (-helix and β-strand)[16].
In the direct observation,gaps are subject to occur in some feasible regions.The gap penalty probably can be tuned in different regions.Therefore, an optimized gap penalty model could be constructed based on this observation.First,the measures of conservation status should be calculated.Second,the gap penalty models based on the highly conserved and less conserved regions could be developed,respectively.The optimized gap penalty model probably can further improve sequence-structure alignment accuracy.
3.3Conclusions
In this manuscript,we reviewed the development history and key algorithms for protein fold recognition methods.In recent years,the evolutionary information generated from iterative PSI-BLAST searches and enlarging NCBI NR database have substantially enhanced the prediction accuracy.The combination of sequence and structural information has also been shown to improve the accuracy of fold recognition. Meanwhile,more and more protein structures are deposited in the Protein Data Bank and it is much easier for a fold recognition method to identify correct templates for a query sequence.Although the development of new fold recognition methods lags behind for several years, the use of this technique is becoming increasingly wider and deeper in the biological community.
Acknowledgements
This work was supported by National Natural Science Foundation of China (NO.31500673), the Education and Science Foundation for Young teachers of Fujian (JA14049), Start-Up Fund of Fuzhou University (XRC-1336), and Science Development Foundation of Fuzhou University (2013-XY-17 and 2014-XY-15).
References
[1] PICOT D, LOLL P J, GARAVITO R M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1[J].Nature,1994,367(6460):243-249.
[2]LEGAULT P, LI J, MOGRIDGE J, et al. NMR structure of the bacteriophage lambda N peptide/boxB RNA complex: recognition of a GNRA fold by an arginine-rich motif [J]. Cell ,1998, 93(2):289-299.
[3]VASSILATIS D K, HOHMANN J G, ZENG H, et al. The G protein-coupled receptor repertoires of human and mouse[J].Proceedings of the National Academy of Science,2003,100(8):4903-4908.
[4]KOONIN E V, WOLF Y I, ARAVIND L. Protein fold recognition using sequence profiles and its application in structural genomics[J]. Advance in Protein Chemistry, 2000,54(54):245-275.
[5]BRAY J E. Target selection for structural genomics based on combining fold recognition and crystallisation prediction methods: application to the human proteome[J]. Journal of Structural & Functional Genomics,2012,13(1):37-46.
[6]SHI J, BLUNDELL T L, MIZUGUCHI K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties[J].Journal of Molecular Biology,2001,310(1):243-257.
[7]KARWATH A, KING R D. Homology induction: the use of machine learning to improve sequence similarity searches[J]. BMC Bioinformatics, 2002,3:11.
[8]BAKER D, SALI A. Protein structure prediction and structural genomics[J]. Science, 2001,294(5540):93-96.
[9]YAN R X, XU D, YANG J Y, et al. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction[J]. Scientific Reports 3, 2013,3:1-9.
[10]ALTSCHUL S F, MADDEN T L, SCHAFFER A A, et al. Gapped blast and psi-blast:a new generation of protein database search programs[J]. Nucleic Acids Research,1997,25:3389-3402.
[11]JAROSZEWSKI L, RYCHLEWSKI L, LI Z, et al. FFAS03: a server for profile-profile sequence alignments[J]. Nucleic Acids Research,2005,33(webserver issue):W284-288.
[12]XU D, JAROSZEWSKI L, LI Z, et al.FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking[J].Bioinformatics,2014,30(5):660-667.
[13]SÖDING J.Protein homology detection by HMM-HMM comparison[J]. Bioinformatics, 2005, 21(7):951-960.
[14]PENG J,XU J.RaptorX: exploiting structure information for protein alignment by statistical inference[J]. Proteins ,2011,79( Suppl 10):161-171.
[15]YAN R X,SI J N, WANG C,et al. DescFold: a web server for protein fold recognition[J]. BMC Bioinformatics, 2009,10:416.
[16]WU S, ZHANG Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information[J]. Proteins-structure Function & Bioinformatics,2008,72(2):547-556.
[17]YANG Y, FARAGGI E, ZHAO H, et al. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates[J]. Bioinformatics, 2011,27(15):2076-2082.
[18]ZHOU H, ZHOU Y. SPARKS 2 and SP3 servers in CASP6[J].Proteins-structure Function & Bioinformatics, 2005,61(s7):152-156.
[19]RUMELHART D E, HINTON G E, WILLIAMS R J. Learning representations by back-propagating errors[J]. Nature, 1986,323:533-536.
[20]BREIMAN L. Random forests[J]. Machine Learning, 2001,45(1):5-32.
[21]BUSUTTIL S, ABELA J, PACE G J.Support vector machines with profile-based kernels for remote protein homology detection[J]. Genome Inform, 2004,15:191-200.
[22]SALI A, BLUNDELL T L.Comparative protein modelling by satisfaction of spatial restraints[J]. Journal of Molecular Biology,1993,234(3):779-815.
[23]PRUITT K D,TATUSOVA T,KLIMKE W,et al.NCBI reference sequences: current status, policy and new initiatives[J]. Nucleic Acids Research ,2009,37(Database issue):D32-D36.
[24]MIZUGUCHI K,DEANE C M,BLUNDELL T L, et al. HOMSTRAD: a database of protein structure alignments for homologous families[J]. Protein Science, 1998,7(11):2469-2471.
[25]JONES D T.GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences[J].Journal of Molecular Biology,1999,287(4):797-815.
[26]XU J,LI M,KIM D,et al.RAPTOR: optimal protein threading by linear programming[J]. Journal of Bioinformatics and Computational Biology, 2003,1(1):95-117.
[27]XU J,PENG J,ZHAO F.Template-based and free modeling by RAPTOR++ in CASP8[J].Proteins,2009,77(Suppl 9):133-137.
[28]MARIANI V,KIEFER F,SCHMIDT T,et al.Assessment of template based protein structure predictions in CASP9[J].Proteins,2011,79(Suppl 10):37-58.
[29]MOULT J,FIDELIS K,KRYSHTAFOVYCH A, et al. Critical assessment of methods of protein structure prediction-Round VIII[J].Proteins,2009,77(Suppl 9):1-4.
[30]FISCHER D,BARRET C,BRYSON K,et al. CAFASP-1:critical assessment of fully automated structure prediction methods[J].Proteins,1999,67(Suppl 3):209-217.
[31]RYCHLEWSKI L,FISCHER D.LiveBench-8: the large-scale, continuous assessment of automated protein structure prediction[J].Protein Science A Publication of the Protein Society,2005,14(1):240-245.
[32]SALWINSKI L,EISENBERG D.Motif-based fold assignment[J].Protein Science A Publication of the Protein Society,2001,10(2):2460-2469.
[33]DONG Q W,WANG X L, LIN L.Application of latent semantic analysis to protein remote homology detection[J]. Bioinformatics, 2006,22(3):285-290.
[34]WU S,ZHANG Y.LOMETS: a local meta-threading-server for protein structure prediction[J]. Nucleic Acids Research, 2007,35(10):3375-3382.
[35]LOBLEY A,SADOWSKI M I,JONES D T.pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination[J]. Bioinformatics ,2009,25(14):1761-1767.
[36]YANG J,YAN R,ROY A,et al.The I-TASSER Suite: protein structure and function prediction[J]. Nat Methods ,2015,12(1):7-8.
[37]MADERA M.Profile Comparer: a program for scoring and aligning profile hidden Markov models[J]. Bioinformatics,2008,24(22):2630-2631.
[38]XU Y, XU D.Protein threading using PROSPECT: design and evaluation[J]. Proteins Structure Function & Bioinformatics,2000,40(3):343-354.
[39]WALLNER B, LARSSON P, ELOFSSON A. Pcons.net: protein structure prediction meta server[J].Nucleic Acids Research, 2008,35(14):W369-374.
[40]LUNDSTROM J,RYCHLEWSKI L, BUJNICKI J, et al.Pcons: a neural-network-based consensus predictor that improves fold recognition[J]. Protein Science A Publication of the Protein Society,2001,10(11):2354-2362.
[41]NEEDLEMAN S B,WUNSCH C D.A general method applicable to the search for similarities in the amino acid sequence of two proteins[J]. Journal of Molecular Biology, 1970,48(3):443-453.
[42]ZHANG W,LIU S,ZHOU Y.SP5: improving protein fold recognition by using torsion angle profiles and profile-based gap penalty model[J]. PLoS One, 2008,3(6):e2325.
A short review of protein fold recognition methods
YAN Renxiang1,WANG Xiaofeng2,XU Weiming1,LIN Juan1, CAI Weiwen1
(1.SchoolofBiologicalSciencesandEngineering,FuzhouUniversity,Fuzhou350108,China;
2.ShanxiNormalUniversityCollegeofMathematicsandComputerScience,LinfenShanxi041004,China)
Abstract:Protein fold recognition method is one of template-based protein three-dimensional structure modeling methods and was elegantly used in many fields of biological sciences.We witnessed the development of a series of novel fold recognition algorithms using different computational techniques in the past ten years.Machine learning and profile-profile alignment are widely used and effective methods.In addition the enlarging Protein Data Bank is one of important factors that substantially enhance the accuracy of prediction.In this paper,we briefly reviewed the state-of-the-art algorithms used in the protein fold recognition and some potential aspects that can be used to improve performance were also discussed.
Keywords:Fold recognition; Sequence alignment; Protein structure prediction
中图分类号:Q51
文献标志码:A
文章编号:1672-5565(2015)04-231-08
doi:10.3969/j.issn.1672-5565.2015.04.05
作者简介:鄢仁祥,男,博士,硕士生导师,研究方向:生物信息学;E-mail: yanrenxiang@fzu.edu.cn.
收稿日期:2015-08-10;修回日期:2015-10-30.
