
牙龈卟啉单胞菌编码基因重注释研究
2015-02-23 07:58徐晓捷计得伟张欣悦张无忌张会雄
生物信息学 2015年4期
关键词:牙周病
徐晓捷,计得伟,张欣悦,张无忌,张会雄*
(1.电子科技大学生命科学与技术学院,神经信息教育部重点实验室,成都610054;
2.电子科技大学信息医学中心,成都 610054;
3. 成都中医药大学针灸推拿学院,成都610000)
牙龈卟啉单胞菌编码基因重注释研究
徐晓捷1,2,计得伟1,2,张欣悦3,张无忌1,2,张会雄1,2*
(1.电子科技大学生命科学与技术学院,神经信息教育部重点实验室,成都610054;
2.电子科技大学信息医学中心,成都 610054;
3. 成都中医药大学针灸推拿学院,成都610000)
摘要:为了确保牙龈卟啉单胞菌生物大分子信息的准确性,对NCBI数据库中的3株牙龈卟啉单胞菌的注释信息进行研究。首先,准备好蛋白质编码与非编码序列正负样本,用基于Z曲线理论的Fisher判别法对正负样本集进行训练,确定一个判断ORF编码或非编码的阈值t0,由阈值作为判别条件来识别所有的ORFs,判断基因片段是否具有编码蛋白质的功能,由此阈值为判别标准排除掉3株牙龈卟啉单胞菌基因组中错误的基因注释信息。然后,用Prodigal基因预测软件对牙龈卟啉单胞菌进行基因预测,基因预测结果与原始功能已知基因进行比对,挑选出具有不同5’终端的ORFs,将这些具有不同5’终端的ORFs与功能已知的基因片段进行比对,找到重叠率小于20%的候选基因。最后,对这些候选基因用Blast进行序列比对找到满足条件的新基因,并为这些新基因添加功能注释信息。基于以上方法共排除了117个非编码的开放式阅读框,并找到了30个NCBI数据库中缺失的编码蛋白质的新基因。
关键词:牙周病;牙龈卟啉单胞菌;基因重注释;新基因
牙周疾病是常见的危害人类牙齿的主要口腔疾病。而牙龈卟啉单胞菌被认为是牙周疾病最重要的致病菌之一,与多种牙周疾病有密切关系。牙周炎是一种慢性口腔疾病,破坏牙齿支持组织,包括胶原蛋白、纤维和骨骼。牙周疾病是由细菌引起的一类感染性疾病,而牙龈卟啉单胞菌(Porphyromonasgingivalis,P.gingivalis)被认为是牙周疾病最重要的致病菌之一。且与成年人、青少年的牙周炎、牙周脓肿、牙槽骨脓肿、牙髓感染以及难治性牙周炎有关。牙龈卟啉单胞菌是牙周病细菌病因学研究的热点[1]。牙龈卟啉单胞菌不仅可以引起发炎,它还与动脉粥样硬化以及肥胖病的发生有关[2-5],且牙龈卟啉单胞菌引起的口腔感染能够通过侵犯主动脉的组织循环加速内皮细胞凋亡[5],造成内皮功能紊乱,许多研究描述了牙周炎导致内皮功能障碍,可通过牙周治疗来改善内皮功能[6]。Curtis等发现,在牙龈卟啉单胞菌W50菌株的55-kDa大外膜上存在着一个由重组活化基因(Recombination activation gene, rag)B编码的相对分子质量为免疫显性表面抗原,与牙周病患者的免疫球蛋白G抗体能否发挥作用有密切关系[7]。通过揭示牙龈卟啉单胞菌生物大分子(如核酸、蛋白质等)的结构,并探索其在遗传信息和细胞信息的传递方式,有助于研究牙龈卟啉单胞菌的致病机理,为研究牙周疾病提供依据。
在基因组公共数据库中已有牙龈卟啉单胞菌基因组的功能注释信息,但是由于很多原因,都有可能造成基因组注释出现有蛋白质功能编码基因被丢弃,或非编码蛋白质功能编码基因被错误标记为功能编码部分的情况出现。可能当时基因组数据库数据量的局限性,或相似基因注释存在错误等,导致基因预测软件会产生一部分错误注释的基因,即非编码的开放式阅读框被预测为编码基因。这就需要研究人员定期对基因组注释信息进行更新。如Bocs等就在26个原核生物全基因组中就发现34%的基因是被错误注释的[8]。还有一种情况是一些真正编码蛋白质的基因,由于种种原因却被丢弃掉了,可以通过一些从头预测的基因查找工具结合基因相似性比对来探测这些基因并为它们添加正确的生物功能信息。近几年,随着基因测序技术的快速发展,尤其是第二代基因测序技术的出现,越来越多的微生物基因组完成了测序,并被上传至公共核苷酸数据库。大量的基因序列数据为人们挖掘更多的生物信息提供了绝佳的机会。与此同时,这也对基因注释信息的准确性提出了更高的要求[9]。如果一个物种的基因组注释出现了错误,那么不仅会影响基于此基因组的后续研究工作,还可能导致与此基因组具有亲缘关系的其他基因组的相关研究工作出现问题,因此为了保证基因注释信息的准确性,需要对数据库中已测序基因组的注释信息进行定期的检查[10]。
针对以上问题,下载了NCBI数据库中最新的牙龈卟啉单胞菌全基因组的注释信息,用基于Z曲线理论的Fisher判别法识别假设基因,排除3株牙龈卟啉单胞菌数据库中被错误注释的假阳性的开放式阅读框(Open reading frames, ORFs),共排除了117个非编码ORFs。增加新基因,即一些真正的能编码蛋白质的基因,由于种种原因被丢弃掉了,需要用基因预测工具并结合基因相似性比对,或通过实验手段探测这些数据库中丢失的基因并为它们添加正确的生物功能注释信息。如Zhou等就通过转录分析和相似性搜索相结合的方法为野油菜黄单胞菌(Xanthomonascampestris)添加了306个新蛋白编码基因[11]。用Prodigal基因预测软件对3株牙龈卟啉单胞菌进行基因预测,把预测基因与原始基因注释信息进行比对,保留重叠率低于20%的预测基因为候选基因,并通过Blast对候选基因进行比对,满足条件的则被认为是要找的新基因,共找到了30个NCBI数据库中缺失的新基因。
1材料和方法
1.1 数据来源
本研究所用的数据主要由两部分组成,一部分是牙龈卟啉单胞菌的全基因组各染色体DNA序列文件(文件扩展名为fna),另一部分是该物种对应的基因在染色体上的位置分布及编码蛋白质功能信息等基因注释数据(文件扩展名为ptt)。这两部分数据都可以从美国国家生物技术信息中心(NCBI)所提供的核酸序列公开数据库(GenBank)的Ftp下载中心(ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/)获得。牙龈卟啉单胞菌全基因组总共包括3个,均是完全测序且在2014年7月之前下载的,它们的全名依次是:PORPHYROMONAS_GINGIVALIS_ATCC_33277_UID58879,PORPTYROMONAS_GINGIVALIS_TDC60_UID67407,PORPHYROMONAS_GINGIVALIS_W83_UID57641,对应的参考序列号为:NC_010729,NC_015571,NC_002950。
基因组注释文件中包含基因片段编码蛋白质功能的描述信息,根据这些描述信息把基因分为三类。第一类是具有明确功能描述的基因,此类基因一般会有确定的基因名称,如gyrB表示DNA旋转酶B亚单位的编码蛋白质。第三类是功能描述为Hypothetical Protein的基因,即在基因注释中不能确定功能信息的假设基因。余下的基因归为第二类基因,一般是在注释文件中具有Family、Putative、Domain等描述词的基因。而第三类基因中还不确定哪些基因真正具有蛋白质编码功能,哪些不具有蛋白质编码功能。因此本文将重点关注第三类基因。
1.2 ORFs判定
要排除基因注释中的非编码ORFs,关键在于建立一个模型和识别方法对所有需要验证的ORFs进行判定。Z-fisher是基于Z曲线理论对假设基因进行检验并排除非编码ORFs[12, 13]。在任意一个基因序列片段或ORF中,把基因序列分为3个相位,第1相位对应第1、4、7、…个碱基所在的位置;第2相位对应第2、5、8、…个碱基所在的位置;第3相位对应第3、6、9、…个碱基所在的位置。根据基因序列的Z变换原理,任意一个基因片段或ORF可由33位空间中的一个点来标识,这33个分量将用作基因编码区的识别变量。具体理论基础和实现过程可参考文献[12-13]。
1.3 去除过注释基因的过程
在重注释过程中首先要排除错误注释的基因信息。基于从头预测的基因预测软件(Gene finder)会产生一部分错误注释的基因,即非编码ORFs被预测为编码基因,这部分基因需要从注释文件中删除。对于本步骤过程的讨论可以参考文献[9]。Zfisher是专业为检查和排除细菌或古细菌非编码ORFs而设计的开源服务系统,可在http://147.8.74.24/Zfisher/获得[9],步骤见图1。

图1 判断第三类基因中的基因序列是否编码蛋白质的流程图Fig.1 The flowchart of judging the gene sequence whetherencoding the protein or not
1.4 查找新基因的过程
在对已测序的基因组进行注释的过程中,为了保证较低的假阳性,一些真正编码蛋白质的基因可能会被遗漏。本研究中使用Blast在线服务中的Blastx程序对所有候选基因的核苷酸序列进行查询。如果一个候选基因的Blast结果同时满足以下4个条件:(1) Evalue<1×10-20,(2) Query Cover>60%,(3) Ident>50%,(4) 候选基因与同源相似基因的长度差<20%,则此候选基因是要找的新基因[9],并为这些新基因添加正确的基因功能信息,具体实现步骤见图2。

图2 用Prodigal基因预测软件对牙龈卟啉单胞菌的基因预测及发现新基因的过程Fig.2 The process of predicting the candidate genesfrom P.gingivalis uesed Prodigal gene predictionsoftware and discovery new genes
2结果与讨论
2.1 基因组大小与基因数量的线性关系
在对牙龈卟啉单胞菌基因组进行重注释之前,先对基因组大小与基因数目之间的关系进行统计分析,本文中用到了2 638个细菌或古细菌的全基因序列及对应的基因注释信息(包括3个牙龈卟啉单胞菌)作为统计分析对象,根据物种的基因组注释信息可以统计出每个染色体的大小及注释的基因数目,并绘制二者的散点分布图(见图3)。图中x轴表示基因组的大小(单位为kb),y轴表示基因数目,从图中可以发现这2 638个细菌或古细菌的基因组大小与基因数目之间具有很强的正相关性(相关系数R=0.994),这说明随着物种基因组的增大,其包含的基因数目也应该随之增多。Mira等也提出,与真核生物相比,大部分原核生物(包括细菌和古细菌等)的编码蛋白质基因紧密的分布在染色体上[14]。此外,由于原核生物中缺少内含子,所以其基因结构比真核生物要简单。可能正是这种紧密的染色体结构以及简单的基因结构,使得细菌或古细菌的基因组大小与基因数目间具有强征相关性。

图3 基因组大小与基因数目关系分布图Fig.3 Linear correlation between genome size and gene number
通过绘制基因组大小与基因数目的线性拟合线(图中黑色虚线),我们发现大部分细菌或古细菌分布在拟合线附近,有部分物种的注释基因数目远多于(或少于)拟合值。针对本文的研究对象,3个牙龈卟啉单胞菌(图中实心圆点),也有类似的规律。由于3个牙龈卟啉单胞菌的基因组大小比较相近(约2 300 K),所以它们在图中几乎分布在同一垂直线上。我们可以发现3个牙龈卟啉单胞菌的注释基因数目分布在拟合性两侧,在基因组大小与基因数量关系方面,这3个牙龈卟啉单胞菌未显示出任何异常。
2.2 去除非编码的ORFs
以P.gingivalisATCC33277为例,基于Fisher判别模型,对正负样本集进行训练,得到判别的阈值,然后比对所有第三类基因,根据阈值判别每一个基因片段是否真正编码蛋白质。在P.gingivalisATCC33277中,有36个假设基因判定为非编码ORFs(见表1)。P.gingivalisW83没有排除的非编码ORFs。P.gingivalisTDC60排除81个非编码ORFs(见表2)。

表1 P.gingivalisATCC33277中排除的36个非编码ORFs基因片段同义号
在一个指定的细菌基因组中,所有的蛋白质编码基因都应该有相似的核苷酸组成结构[15],也就是说P.gingivalisATCC33277中的假设基因需要与其功能已知基因具有相似的核苷酸结构,否则将被判定为非编码ORFs。相似性核苷酸结构的判定,正是通过判别模型来确定的,在判别模型中会根据33个识别变量确定此核苷酸序列的阈值,通过此阈值判定是否编码蛋白质,排除这36个假设基因正是基于此判别方法[12]。下图是P.gingivalisATCC33277菌株1 125个功能已知基因(蓝色*圆点标记)和36个非编码ORFs(黑色*圆点标记)的核苷酸散点分布图(见图4)。注:*图中颜色标注见电子版(http://swxxx.alljournals.cn/index.aspx)(2015年第4期)。
表2 P.gingivalisTDC60中排除的81个非编码ORFs基因片段同义号
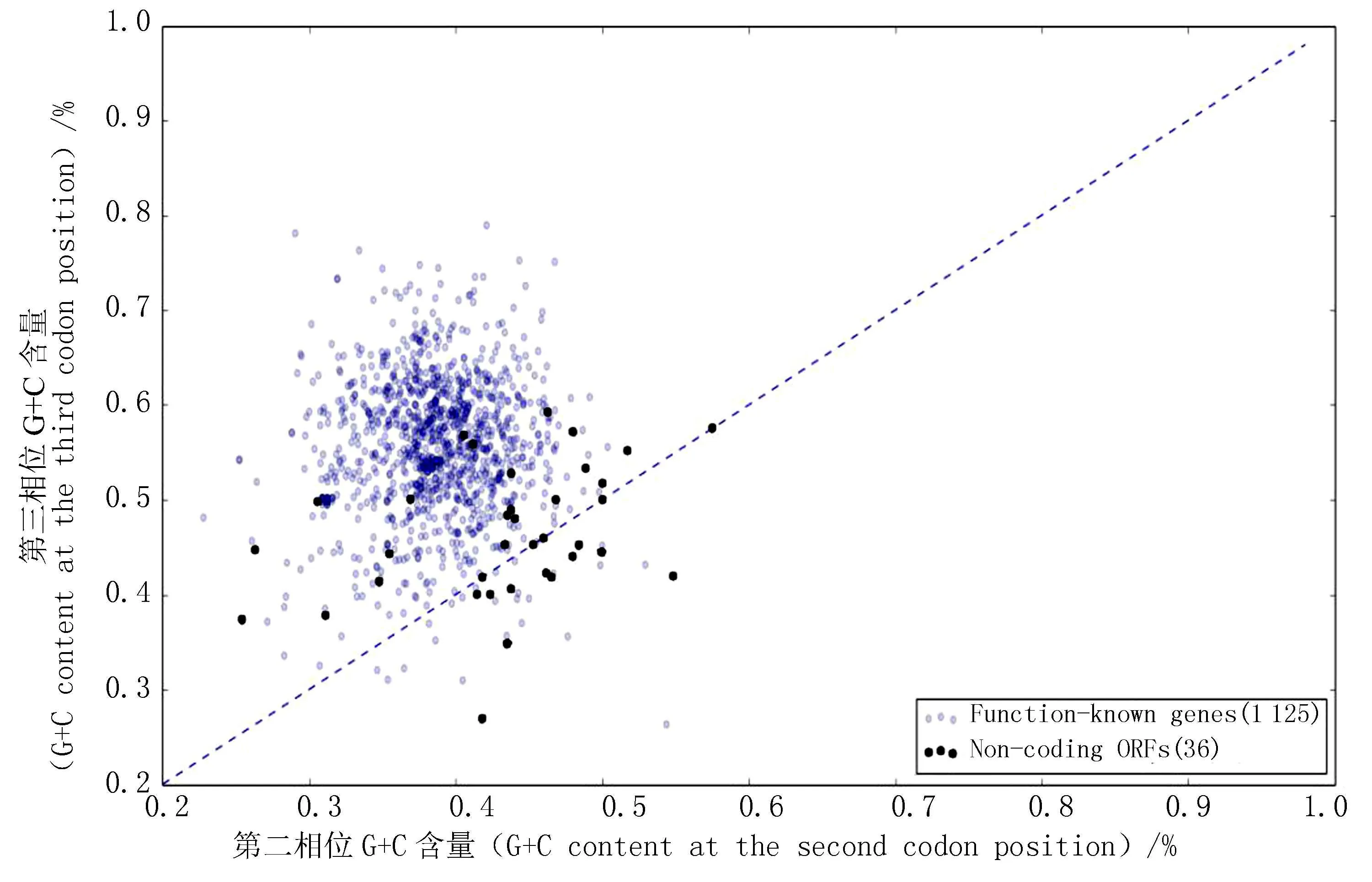
图4 P.gingivalis ATCC33277全基因组G+C含量散点分布图Fig.4 P.gingivalis ATCC33277genome G+C content scatter distribution
从图中可以观察到绝大部分的功能已知基因与非编码ORFs相分离。而且几乎所有的功能已知基因都位于45度对角线上方,这说明其第二相位G+C含量要低于第三相位G+C含量。而36个非编码ORFs中绝大部分分布在45度对角线附近,这表明其第二、三相位的G+C含量基本相同。由此可见编码功能蛋白质将会影响基因的核苷酸结构分布[13, 16, 17]。因此,由于这36个假设基因与功能已知基因具有不同的核苷酸结构,在判别模型中得到的判别值不满足编码蛋白质的Z曲线阈值,导致其被排除为非编码ORFs。
2.3 找到新基因,添加功能信息
使用Blast在线服务对所有候选基因的核苷酸序列进行查询。如果一个候选基因的Blast结果同时满足4个条件:(1) Evalue<1×10-20,(2) Query Cover>60%,(3) Ident>50%,(4)候选基因与同源相似基因的长度差<20%,我们就认为此候选基因是要找的新基因。通过以上方法,从3株牙龈卟啉单胞菌中分别找到了不同数量的新基因。在P.gingivalisTDC60中找到了6个新基因(见表3)。这6个新基因的基因位置与原注释中的基因位置重叠率很低,全部小于0.05%,其中还包括5个重叠率几乎为0的新基因,即原注释信息中几乎没有覆盖到的基因。根据同源基因的功能描述确定新基因的功能信息,同时这6个新基因也被赋予各自同源基因的功能注释信息,如新基因348 817-348 960(+)则被注释为转座酶(Transposase)。
表4和表5分别是P.gingivalisATCC33277和P.gingivalisW83中发现的新基因以及其相应的功能注释信息。

表3 P.gingivalisTDC60中发现的6个新基因信息

表4 P.gingivalisATCC33277中发现的5个新基因信息
表5 P.gingivalisW83中发现的19个新基因信息
3结论与展望
基因组重注释方法是根据Fisher判别法识别3株牙龈卟啉单胞菌所有第三类基因(假设基因),判定基因片段是否具有编码蛋白质功能。基于此方法从3株牙龈卟啉单胞菌中共排除了117个非编码ORFs。对牙龈卟啉单胞菌使用基于从头预测方法的基因识别工具Prodigal查找候选新基因,并以最新的基因数据库为基础进行Blast在线相似性比对查找同源基因,最后根据设定的参数阈值对结果进行过滤筛选,确定满足条件的新基因并添加对应的基因功能信息,在本文中为牙龈卟啉单胞菌共添加了30个新基因。经过本文的重注释,可能仍然还存在未排除的非编码ORFs和未找到的新基因。为保证结果的可靠性,使用特异性较低的方法排除非编码ORFs(低至54%),同时在查找新基因的过程中只保留高相似度的结果(高达99%)。随着这两个参数的变化,发现新基因的数量和排除的非编码基因的ORF的数量都有可能会变化。本研究中,用Prodigal基因预测软件识别基因位置,后续可以扩展使用更多其他的基因预测软件对假设基因进行验证,以确保结果的可靠性。
参考文献
[1]黄定明, 吴亚菲. 牙龈卟啉单胞菌的分型及其致病作用[J].国外医学: 口腔医学分册,2002, 29(4): 213-215.
HUANG Dingming, WU Yafei. Typing and pathogenic role of porphyromonas gingivalis aeromonas[J]. Foreign Medical: Stomatology Volume, 2002,29(4): 213-215.
[2]SHAH P K. Plaque disruption and thrombosis: potential role of inflammation and infection[J].Cardiology in Review, 2000,8(1): 31-39.
[3]KUVIN J T,KIMMELSTIEL C D.Infectious causes of atherosclerosis[J].American Heart Journal,1999,137(2):216-226.
[4]CAI Y, KOBAYASHI R, HASHIZUME-TAKIZAWA T, et al. Porphyromonas gingivalis infection enhances Th17 responses for development of atherosclerosis[J].Archives of Oral Biology, 2014, 59(11): 1183-1191.
[5]AO M , MIYAUCHI M , INUBUSHI T, et al. Infection with porphyromonas gingivalis exacerbates endothelial Injury in obese mice[J].PloS One,2014,9(10): e110519-e110519.
[6]GURAV A N. The implication of periodontitis in vascular endothelial dysfunction[J].European Journal Of Clinical Investigation, 2014,44(10): 1000-1009.
[7]HANLEY S A , ADUSE-OPOKU J , CURTIS M A . A 55-Kilodalton immunodominant antigen of porphyromonas gingivalis W50 Has arisen via horizontal gene transfer[J].Infection and Immunity, 1999, 67(3): 1157-1171.
[8]BOCS S,DANCHIN A,MÉDIGUE C.Re-annotation of genome microbial coding-sequences:finding new genes and inaccurately annotated genes[J].BMC Bioinformatics,2002,3(1):1-10.
[9]GUO F B , XIONG L , TENG L , et al. Re-annotation of protein-coding genes in 10 complete genomes of Neisseriaceae family by combining similarity-based and composition-based methods[J].DNA Research,2013,20(3):273-286.
[10]CAMUS J C,PRYOR M J ,MéDIGUE C,et al.Re-annotation of the genome sequence of mycobacterium tuberculosis H37Rv[J].Microbiology,2002,148(10):2967-2973.
[11]ZHOU L,VORHÖLTER F J,HE Y Q ,et al.Gene discovery by genome-wide CDS re-prediction and microarray-based transcriptional analysisinphytopathogenXanthomonascampestris[J].BMC Genomics,2011, 12(1):359.
[12]ZHANG C T , ZHANG R . Analysis of distribution of bases in the coding sequences by a diagrammatic technique[J].Nucleic Acids Research, 1991, 19(22): 6313-6317.
[13]ZHANG C T , CHOU K C . A graphic approach to analyzing codon usage in 1562 Escherichia coli protein coding sequences[J].Journal of Molecular Biology,1994, 238(1): 1-8.
[14]MIRA A , OCHMAN H , MORAN N A . Deletional bias and the evolution of bacterial genomes[J].Trends Genet,2001, 17(10): 589-596.
[15]ZHANG C T , WANG J . Recognition of protein coding genes in the yeast genome at better than 95% accuracy based on the Z curve[J].Nucleic Acids Research,2000, 28(14): 2804-2814.
[16]GUO F B. The distribution patterns of bases of protein-coding genes, non-coding ORFs, and intergenic sequences in pseudomonas aeruginosa PA01 genome and its implications[J].Journal of Biomolecular Structure and Dynamics,2007,25(2):127-133.
[17]CHEN L L , ZHANG C T . Seven GC-rich microbial genomes adopt similar codon usage patterns regardless of their phylogenetic lineages[J].Biochemical And Biophysical Research Communications,2003,306(1): 310-317.
Re-annotation ofPorphyromonasgingivaliscoding-sequences
XU Xiaojie1,2, JI Dewei1,2, ZHANG Xinyue3, ZHANG Wuji1,2, ZHANG Huixiong1,2*
(1.SchoolofLifeScienceandTechnology,KeyLabofNeuroinformationofMinistryofEducation,
UniversityofElectronicScienceandTechnology(UESTC),Chengdu610054,China;
2.MedicalInformaticsCenter,UESTC,Chengdu610054,China;
3.SchoolofAcupunctureandMassage,ChengduUniversityofTCM,Chengdu610054,China)
Abstract:To ensure accuracy ofP.gingivalisbiological macromolecules information,we investigated the annotations of the 3P.gingivalisbased on NCBI database. Firstly, we prepared protein-coding and non-coding sequences as positive and negative samples,respectively,and used Fisher Discriminant which was designed based on Z curve theory to determine the threshold t0,which was used as the criterion to determine whether the gene encoding the protein or not. We firstly excluded the wrong annotation information from three stains ofP.gingivalisbased on the threshold. Secondly, theP.gingivaliswere predicted with the prodigal gene prediction software. We used the predicted genes compared to the original known-function genes and selected the ORFs with different 5’terminals, identified the candidate genes with overlapping rate of less than 20% from the ORFs with different 5’terminals.Finally, we used the sequence alignment software Blast to find the candidate genes that meet the conditions. We excluded 117 non-coding open reading frames, and found 30 new protein-coding genes that were not annotated in the NCBI database.
Keywords:Periodontal disease;Porphyromonasgingivalis; Re-annotation; New genes
中图分类号:Q343.1+2
文献标志码:A
文章编号:1672-5565(2015)04-205-07
doi:10.3969/j.issn.1672-5565.2015.04.01
作者简介:徐晓捷,女,硕士研究生,研究方向:生物医学工程;E-mail:517170490@qq.com.*通信作者:张会雄,副教授,研究方向:移动互联与公众健康;E-mail:940351908@qq.com.
基金项目:中央高校基本科研业务费(ZYGX2013J100);2014年非全日制专业学位研究生教研教改项目(ZY2014009)。
收稿日期:2015-07-19;修回日期:2015-09-10.
猜你喜欢
中华养生保健(2020年7期)2020-11-16
山西医科大学学报(2020年7期)2020-08-12
江苏卫生保健(2020年12期)2020-02-13
中国实用乡村医生杂志(2020年4期)2020-01-12
中华老年口腔医学杂志(2016年4期)2017-01-15
妇女之友(2016年8期)2016-10-21
中华胃食管反流病电子杂志(2016年3期)2016-10-18
中国卫生标准管理(2015年16期)2016-01-20
食管疾病(2015年3期)2015-12-05
中国医疗美容(2015年2期)2015-07-19
