
藤黄中藤黄酸B的制备分离方法优选及结构确证
2015-02-19 02:02贾步云彭代银李珊珊胡雪瑞朱光宇陈卫东
安徽中医药大学学报 2015年1期
贾步云,彭代银,李珊珊,胡雪瑞,朱光宇,2,陈卫东
(1.安徽中医药大学药学院,安徽 合肥 230012; 2.安徽省马鞍山市中心医院,安徽 马鞍山 243000)
藤黄中藤黄酸B的制备分离方法优选及结构确证
贾步云1,彭代银1,李珊珊1,胡雪瑞1,朱光宇1,2,陈卫东1
(1.安徽中医药大学药学院,安徽 合肥230012; 2.安徽省马鞍山市中心医院,安徽 马鞍山243000)
[摘要]目的建立一种高效且重现性好的藤黄酸B的制备分离方法,并对分离产物进行结构确证。方法采用乙醇超声提取方式对藤黄药材进行初步提取,用中压制备色谱和高压制备色谱相结合的方式对藤黄乙醇提取物中藤黄酸B进行分离和纯化,并采用正交设计对提取和分离工艺进行优化。采用红外光谱、质谱、1H-核磁共振谱、13C-核磁共振谱对分离得到的样品进行结构鉴定。结果优选的分离方法较其他方法简便且高效可控,分离得到的样品通过结构鉴定,确定为藤黄酸B且纯度可达到99%以上。结论所建立的藤黄酸B的提取分离方法具有高效、简便且重现性好的特点。同时,本研究提供了藤黄酸B相对完整的谱图信息,并确证了所分离藤黄酸B的结构。
[关键词]藤黄;藤黄酸;有效成分分离;结构确证
中药藤黄是藤黄科植物藤黄(GarciniahanburyiHook. f)的干燥树脂,具有攻毒蚀疮、破血散结、止血、杀虫的功效[1],临床上用于治疗痈疽肿毒、顽癣恶疮、创伤出血及烫伤[2]。现代研究发现藤黄中具有多种抗癌有效成分,而目前的研究热点主要集中在藤黄酸(gambogic acid, GA)和新藤黄酸(gambogenic acid, GNA)这两种化合物。通过查阅相关文献,发现藤黄酸B(morellic acid,MA)也具有较强的抗癌活性,MA对于人白血病K5652及K562/s细胞株有很好的抑制作用[3];同时,MA也具有一定的抗菌活性[4]。目前对于MA分离纯化的相关研究较少,且步骤相对较为繁琐[3-5]。本研究采用中压制备液相色谱以及高压制备色谱从藤黄的乙醇提取物中分离纯化得到MA,优选出一种简便且重现性好的高纯度MA的提取分离方法,并对分离得到的MA样品进行结构确证,为MA的进一步研究奠定基础。
1仪器与材料
1.1仪器中压制备液相色谱:包括TAUTOGrad200型中压二元梯度泵TAUTOTBD2000型紫外检测器:上海同田生物技术有限公司;中高压玻璃制备色谱塔310 mm×100 mm,填料ODS C1830~50 μm:苏州汇通色谱分离纯化有限公司;N2000色谱工作站:浙江大学智达信息工程有限公司;Waters 2545高压制备色谱系统:美国Waters公司; AB SCIEX QTRAP 4500系统:美国AB公司;超导傅立叶数字化核磁共振谱仪:瑞士布鲁克公司;岛津LC-15C高效液相色谱仪、紫外可见分光光度计、红外光谱仪:日本岛津公司;RE-2000B型旋转蒸发器:上海亚荣生化仪器厂;AB125-S型十万分之一分析天平:德国梅特勒公司。
1.2材料甲醇(分析纯):安徽省安庆时联特种溶剂股份有限公司;甲醇(色谱纯):上海星可高纯溶剂有限公司;甲酸、无水乙醇、乙酸乙酯、丙酮、石油醚:均为分析纯,江苏强盛功能化学股份有限公司;藤黄:批号 201103-1,购自安徽亳州药材市场,经安徽中医药大学王效山教授鉴定,为藤黄科植物藤黄(GarciniahanburyiHook. f.)的干燥树脂;MA对照品由安徽中医药大学科研实验中心提供,纯度≥98%。
2MA分析方法的建立
2.1色谱条件岛津LC-15C高效液相色谱仪(包括LC-15C高压输液泵、SPD-15C紫外检测器);色谱柱:COSMOSIL C18柱(4.6 mm×250 mm,5 μm);流动相:甲醇∶水(90∶10);柱温:30 ℃;进样量20 μL;流速:1.0 mL/min;检测波长:360 nm。MA色谱图见图1。
2.2对照品溶液的配制精密称取MA对照品10.0 mg,置于50 mL容量瓶中,加甲醇定容至刻度线,即得200 μg/mL的对照品溶液,于4 ℃冰箱保存。
2.3供试品溶液的配制精密称取MA样品10.0 mg,置于50 mL容量瓶中,加甲醇定容至刻度线,即得200 μg/mL的对照品溶液,于4 ℃冰箱保存。
2.4线性关系考察精密移取0.025、0.25、0.5、1、1.5、2、4、8 mL的对照品溶液,置于50 mL容量瓶中,加甲醇定容至刻度,配制成0.1、1、2、4、6、8、16、32 μg/mL的标准系列浓度,摇匀及过滤后,进样分析,以峰面积(y)对MA的质量浓度(x)进行线性回归,回归方程:y=32 080x-589.34(r=0.999 8),表明MA在0.1~32 μg/mL浓度范围内线性良好。
2.5稳定性试验取供试品溶液分别于0、0.5、1、2、3、4、5 h进样分析,结果MA峰面积的RSD为1.22%,表明MA的供试品溶液在5 h内稳定。
2.6精密度试验取标准品溶液200 μg/mL,重复进样5次,测定MA峰面积,计算MA峰面积的RSD值为0.71%。
2.7重复性试验平行制备6份MA供试品溶液,进样分析,计算MA峰面积RSD值为1.48%。
2.8加样回收率试验制备质量浓度分别为20、50、100 μg/mL的供试品溶液各3份(共9份),每组分别按供试品量的80%、100%、120%精密加入对照品溶液,用流动相配成合适浓度,进样分析,计算得平均加样回收率为99.74%,RSD为1.04%。
3分离与纯化
3.1藤黄总酸的提取
3.1.1提取方法取藤黄100 g,粉碎后用乙醇超声提取,提取后静置30 min,待溶液分层后,取上层液进行抽滤。取滤液在50 ℃条件下减压蒸馏至浓稠状,取出黏稠的液体倒入蒸发皿内,在55 ℃下真空干燥5~6 h,待完全膨胀疏松后,取出产物进行称量,即得藤黄总酸。
3.1.2提取方法优化采用正交设计对于MA的中压制备色谱分离方法进行优选,通过预实验选取了乙醇浓度(A)、乙醇用量(B)、提取时间(C)、提取次数(D)为考察因素,以MA的得率为考察指标,因素水平表如表1所示。

表1 正交试验因素水平表
3.1.3优化结果与分析在平行操作条件下,对按照L9(34)正交试验提取的9份干浸膏处理后分别进样,测定MA的含量。正交试验结果及方差分析见表2、表3。根据表2直观分析极差大小可知,影响MA提取因素的主次顺序为:A>D>B>C;从方差分析可见,A因素的主效应有统计学意义。综合考虑二者,藤黄总酸提取的最佳工艺条件为:A1B2C3D3,即提取工艺为95%乙醇,6倍量,每次提取3 h,提取3次。
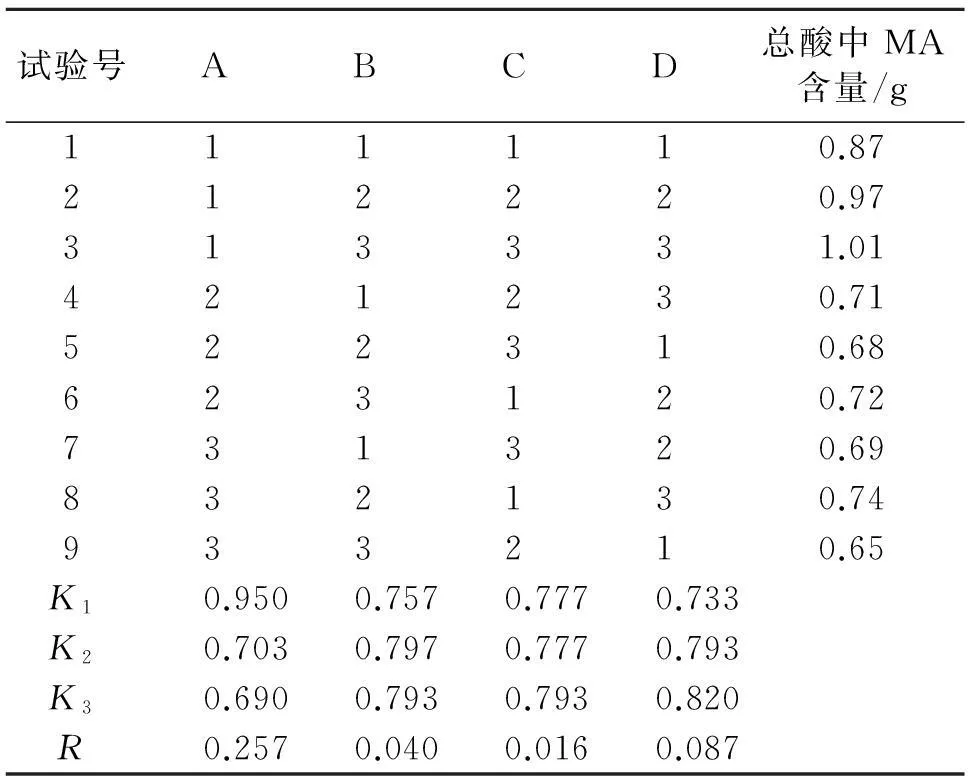
表2 正交试验结果
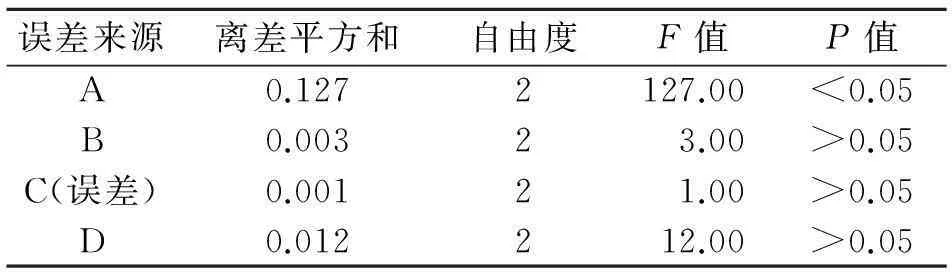
表3 L9(34)正交试验的方差分析结果
注:F0.05(2,2)=19.00。
3.1.4醇提优化工艺验证试验按照优化后的工艺进行试验,测得3批干浸膏中MA的平均含量为0.99 g,RSD值为1.5%,表明该提取工艺稳定可行。
3.2MA的中压液相色谱分离
3.2.1样品前处理称取一定量藤黄总酸粉末,加入无水乙醇10 mL,在40 ℃条件下超声20 min后呈黏稠状,备用。
3.2.2分离方法取下中高压玻璃制备色谱塔预柱的上盖,将“2.2.1”项下所述试样倒入预柱的填料中并搅拌均匀,在拌样硅胶顶端撒上防冲硅胶后拧紧上盖,打开中压制备液相色谱。为防止流速过快将样品冲散,应逐渐提高流动相流速。按照色谱工作站上的色谱图(图2)接取保留时间125.4~185.7 min的色谱峰所对应的洗脱液,55 ℃减压蒸馏,待溶液刚刚饱和时取下,在室温下放置2~3 d自然结晶,使用微孔滤膜进行抽滤,得MA粗品。
3.2.3分离工艺优选采用正交设计对于MA的中压制备色谱分离方法进行优选,选取上样量(A)、流动相比例(B)以及流速(C)为考察因素,以MA的得率为考察指标,因素水平表见表4。

表4 正交试验因素水平表
3.2.4优选结果及分析在平行操作条件下,按照L9(34)正交试验进行9次MA的中压制备色谱分离,测定MA的得率。正交试验结果及方差分析见表5、表6。由表2极差大小的直观分析可知,影响MA提取因素的主次顺序为:A>B>C;从方差分析可见,A因素的主效应具有统计学意义。综合考虑二者,藤黄总酸提取的最佳工艺条件为A2B2C2,即MA中压制备色谱分离工艺为流动相甲醇和水的比例为80∶20,上样量35 g,流速150 mL/min。

表5 L9(34)正交试验结果

表6 L9(34)正交试验的方差分析结果
注:F0.05(2,2)=19.00。
3.2.5中压制备色谱分离优化工艺验证实验按照优化后的工艺进行实验,测得3批分离所得MA粗品的平均得率为80.89%,RSD值为1.6%,表明该分离工艺稳定可行。
3.3MA的高压制备色谱纯化
3.3.1样品处理取MA粗品1 g逐渐加入甲醇溶解成饱和溶液后,用0.45 μm滤膜过滤,备用。
3.3.2分离纯化取“2.3.1”项下样品,通过Waters 2545高压制备色谱系统进行纯化,色谱条件为色谱柱:SunFire prep C18(32 mm×250 mm,5 μm);流动相:甲醇∶水(V/V)90∶10;流速:15 mL/min;进样量:1 mL;检测波长:360 nm;洗脱液自动接收程序:按照最小接收峰面积150 000接收洗脱液;收集保留时间为33.00~34.00 min洗脱液,并蒸干溶剂,即得纯化后的MA样品。见图3。
3.4理化性质及鉴别分离得到的MA为橙黄色粉末,熔点温度为108~109 ℃。分别采用氯仿-甲醇、石油醚-乙酸乙酯及石油醚-丙酮3种不同的展开剂体系进行薄层鉴别,均为单一斑点。本研究采用高效液相色谱法面积归一化法计算分离得到产物的纯度,结果表明纯度大于99%,符合结构鉴定的要求。
3.5工艺流程图见图4。
4MA的结构确证
4.1质谱取5 μg/mL的MA样品,于质谱中分析,样品的电喷雾质谱图给出其准分子离子峰m/z为561.1(M+),其分子量约为561。同时,图谱也给出了其主要碎片峰,分别为:505.0(C29H29O8),477.1(C28H29O7),459.0(C28H27O6),441.0(C28H25O5),418.9(C26H27O5),378.8(C23H23O5),337.8(C19H14O6),229.1(C13H9O4)。将上述质谱数据与文献[6]中MA的电喷雾质谱数据进行比对,数据一致。
4.2红外光谱MA样品的红外光谱图中3 080 cm-1处强吸收峰为酚羟基伸缩振动。2 976、2 928 cm-1处强吸收峰分别为甲基和亚甲基结构的伸缩振动,1 435 cm-1处的强吸收峰为甲基和亚甲基结构的不对称面内弯曲振动,1 364 cm-1处的强吸收峰为甲基的对称面内弯曲振动,1 384 cm-1处近似等高双峰为偕二甲基的特征吸收。3 360 cm-1处强吸收峰为羧酸上羟基的伸缩振动,1 738 cm-1处强吸收峰为羧酸上羰基的伸缩振动。1 690 cm-1处强吸收峰为酮羰基的伸缩振动。1 651、1 593、1 435 cm-1处的强吸收峰为芳环骨架的伸缩振动。1 302 cm-1处的强吸收峰以及1 166 cm-1处的弱吸收峰为酚羟基碳氧键的伸缩振动。1 247 cm-1处的强吸收峰为芳香醚碳氧键的伸缩振动。上述结果表明:MA样品化学结构中存在各官能团红外光谱特征与MA的化学结构红外吸收理论值基本一致。
4.3核磁共振光谱用氘代甲醇溶解试样,采用四甲基硅烷作为内标,测定MA的核磁共振氢谱为:1H-NMR(MeOD, 400 MH)δ12.75(1H,s), 7.56(1H, d,J=6.8 Hz), 6.52(1H, d,J=10.0 Hz), 6.07(1H, t,J=7.0 Hz), 5.44(1H, d,J=10.0 Hz), 5.02(1H, d,J=6.0 Hz), 3.45(1H, dd,J=6.4 Hz, 4.8 Hz), 3.32(2H, m), 3.07(2H, m), 3.01(2H, m), 2.47(1H, d,J=9.6 Hz), 2.31(1H, dd,J=13.6 Hz,J=4.5 Hz), 1.71(3H, s), 1.69(3H, s), 1.67(3H, s), 1.61(3H, s), 1.38(3H, s), 1.37(3H, s), 1.26(3H, s)。
用氘代甲醇溶解试样,采用四甲基硅烷作为内标,测得MA的核磁共振碳谱如下:
13C-NMR(MeOD, 100 MHz)δ204.59,180.14,170.66,162.64,157.47,157.05,137.54,135.14,132.66,130.86,127.73,126.05,122.23,116.87,109.75,104.57,101.28,90.82,83.16,79.23,50.74,47.10,30.91,29.56,28.37,28.22,28.18,26.48,26.43,21.87,19.04,18.91,12.40。
将上述核磁共振光谱数据与文献[7]中MA的数据进行比对,数据一致。
5结果
本研究将中压制备色谱与高压制备色谱技术相结合,建立了一种高纯度MA的提取分离纯化方法,优选出的最佳工艺为:6倍量95%乙醇超声提取3次,每次提取3 h得到藤黄总酸;采用中压制备液相,在流动相甲醇∶水(80∶20),上样量35 g,流速为150 mL/min条件下从藤黄总酸中分离得到MA粗品,最后采用高压液相色谱进行纯化。分离得到的样品用紫外光谱、红外光谱、质谱、1H-核磁共振谱、13C-核磁共振谱进行结构鉴定,并与相关文献数据进行比对,结果表明,样品确定为MA(见图5),经高效液相色谱法面积归一化法计算纯度可达99%以上。
6讨论
中药藤黄中的化合物绝大部分都具有桥环结构,化学结构相似,因此对于这些化合物的分离存在一定难度。以往对于藤黄中化合物分离方法的研究往往是多种成分同时分离,需要反复过柱纯化,步骤相对繁琐,且重现性不好,不利于这些活性成分的深入研究。本研究所建立的提取分离方法针对MA这一单体化合物,采用现代的制备分离仪器,不仅提高分离效率,且具有良好的重现性和可操作性,为MA的进一步研究打下基础。由于本实验采用的分离纯化实验的规模较大,因此在MA的工业化生产方面也有一定的应用前景。同时,本研究还提供了MA相对完整的光谱谱图数据,完善了MA的结构信息,对MA及同类化合物的结构鉴定以及质量控制有一定的价值。
参考文献:
[1]肖国丽,赵学军,刘卫海,等.新藤黄酸对S180细胞株的体内外抗肿瘤作用[J].中国实验方剂学杂志,2012,18(13):193-197.
[2]窦娟,文红梅,郁红礼,等.藤黄炮制品对大鼠肠道组织病理学研究[J].中国实验方剂学杂志,2013,19(5):279-282.
[3]Han QB, Wang YL, Yang L, et al. Cytotoxic polyprenylated xanthones from the resin ofGarciniahanburyi[J]. Chem Pharm Bull, 2006, 54(2):265-267.
[4]Sukpondma Y, Rukachaisirikul V, Phongpaichit S. Antibacterial caged-tetraprenylated xanthones from the fruits ofGarciniahanburyi[J]. Chem Pharm Bull, 2005,53(7):850-852.
[5]贾明美,寿清耀,谭青,等.藤黄化学成分的研究[J].化学学报,2008,66(22):2513-2517.
[6]Zhou Y, Liu X, Yang J, et al. Analysis of caged xanthones from the resin ofGarciniahanburyiusing ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry[J]. Anal Chim Acta, 2008(1-2),629:104-118.
[7]Lee Senbin. Fractionated products obtained from gamboge, and the medical uses of the same:United States, 0305784[P]. 2011-11-15.
Optimization of Extraction and Isolation of Morellic Acid inGarciniahanburyiand Its Structure Confirmation
JIABu-yun1,PENGDai-yin1,LIShan-shan1,HUXue-rui1,ZHUGuang-yu1,2,CHENWei-dong1
(1.SchoolofPharmacy,AnhuiUniversityofChineseMedicine,AnhuiHefei230012,China;2.Ma’anshanCentralHospital,AnhuiMa’anshan243000,China)
[Abstract]ObjectiveTo establish an efficient and repeatable method for isolation of morellic acid and to confirm the structure of isolated product. MethodsGarcinia hanburyi was subjected to ethanol ultrasonic extraction, and the ethanol extract was treated by a combination of medium-pressure preparative chromatography and high-pressure preparative chromatography to isolate and purify morellic acid. An orthogonal design was used to optimize the extraction and isolation process. Also, infrared spectrometry, mass spectrometry,1H-nuclear magnetic resonance (NMR), and13C-NMR were used for the structural identification of isolated sample. ResultsThe optimized isolation method was simple and controllable. The isolated sample was structurally identified as morellic acid with a purity over 99%. ConclusionThe established extraction and isolation method for morellic acid is efficient, simple, and repeatable. This study also provides relatively full spectral information on morellic acid and confirms the structure of isolated morellic acid.
[Key words]Garcinia hanburyi; morellic acid; isolation; structure confirmation
收稿日期:(2014-08-01;编辑:张倩)
通信作者:陈卫东,anzhongdong@126.com
作者简介:贾步云(1989-),男,硕士研究生
基金项目:国家科技部重大新药创制专项(2009ZX09103-399)
[中图分类号]R284[DOI]10.3969/j.issn.2095-7246.2015.01.024
