
基于序列预测蛋白质和RNA的相互作用
2015-02-18 01:30:00李享云
大连交通大学学报 2015年4期
汪 颖,李享云
(大连交通大学 理学院,辽宁 大连 116028)*
0 引言
蛋白质与RNA的相互作用在许多生理过程中起着重要的作用,RNA参与很多基本的细胞生理过程,如携带来自DNA的遗传信息,参与形成核糖体、拼接体、端粒酶等许多核酸蛋白颗粒的结构,有些RNA还具有酶活性等,几乎所有的RNA生物功能的发挥都需要蛋白质的共同作用.蛋白质和RNA相互作用的研究为最终探明RNA和蛋白质相互作用的分子机制,从本质上认识相关细胞生理过程起着不可忽视的重要作用.采用实验的方法预测蛋白质和RNA的相互作用有很大的局限性,或因实验步骤过多,既耗时又费力,也增加了实验结果的不稳定性.因此,利用计算的方法预测蛋白质和RNA相互作用成为当前的一大趋势.
近年来,由于RNA本身的复杂性导致蛋白质和RNA相互作用的研究一直处于滞后状态.但是随着实验获取的RNA数据以及蛋白质和RNA复合物数据的增加,蛋白质和RNA相互作用的预测方法研究成为目前非常紧迫的一项重要课题.2011 年,Pancaldi和Bahler[1]首次提出了一种预测蛋白质和RNA相互作用的方法,选取100多种显著性较高的特征(包括Gene Ontology条款,基因和蛋白质的物理性质,mRNA性质,蛋白质的二级结构以及基因的相互作用genetic interactions等)构建特征向量.然而,由于该文中用到的特征种类较多,有些特征不易获取,所以这种方法具有一定的局限性.同年,Bellucci等人[2]提出一种新的预测蛋白质-RNA相互作用的方法catRAPID,考虑存在于氨基酸链和核苷酸链中的几乎所有关联,从中选取了倾向性较高的二级结构、氢键和范德华这三种性质,并基于此计算每个RNA和蛋白质对的相互作用倾向性,用于预测蛋白质和NRA的相互作用.以上两种方法均考虑了蛋白质和RNA多种性质特征.对于现有的蛋白质-RNA数据而言,都有着一定的局限性.于是在2011年和2013年,文献[3-4]主要基于蛋白质和RNA序列信息,即氨基酸和核苷酸的成分特征,构建机器学习模型.在研究[4]中,基于蛋白质序列中氨基酸组成成分以及RNA序列中核苷酸组成成分,通过特征选取的方法提取有效特征构建向量,从而构建预测模型.通过对多组数据的预测,证实了特征选取方法以及预测模型的有效性.但是,特征选取方法也存在一个弊端,即被选取的特征在某种程度上依赖于样本数据.
本文基于蛋白质和RNA序列,提出了一种新的预测蛋白质-RNA相互作用的方法.本文只考虑了氨基酸三联体和核苷酸的组成成分,利用其成分比率以及氨基酸三联体-核苷酸相互作用倾向性构建了一种新的用于衡量蛋白质和RNA序列对个体的三联体-核苷酸倾向性度量,并利用该倾向性以及氨基酸三联体和核苷酸的成分特征构建支持向量机(support vector machine,SVM)模型,预测其相互作用.
1 预测模型和算法
1.1 氨基酸三联体-核苷酸的相互作用倾向性
氨基酸三联体[5]指的是三个连续的氨基酸构成的一个整体.蛋白质序列中共有20种氨基酸,则三联体的总个数为20×20×20=8 000个;RNA序列中有4种核苷酸,因而共有8 000×4=32 000个氨基酸三联体-核苷酸组合.在文献[6]中,针对一组来自于蛋白质数据库(PDB)的3149个具有相互作用的蛋白质-RNA对,利用文献[6]中氨基酸三联体-核苷酸的相互作用倾向性度量,即文献[6]中式(1),计算得到了氨基酸三联体-核苷酸的相互作用倾向性值,见附表1.附表1中出示了32 000个三联体-核苷酸组合的倾向性值,这些值是针对来自PDB的3149个蛋白质-RNA序列对这个整体数据集而言,氨基酸三联体-核苷酸的相互作用倾向性,在这里称之为整体三联体-核苷酸倾向性.
蛋白质和RNA是否相互作用主要取决于氨基酸和核苷酸位点的结合上.因此本文试图利用氨基酸三联体-核苷酸的倾向性这一性质构建特征向量.为了更好地度量每一对蛋白质-RNA序列个体对中三联体-核苷酸的相互作用倾向性,我们重新定义了一个权重倾向性度量,也可称为个体三联体-核苷酸倾向性度量,如下:

其中,P表示蛋白质序列;R表示RNA序列;t表示蛋白质序列P中的氨基酸三联体;b表示RNA序列R中的核苷酸;Nt,Nb分别表示蛋白质序列P中氨基酸三联体t的数量和RNA序列中核苷酸b的数量;NP,NR表示蛋白质序列P中所有氨基酸三联体的数量和RNA序列中所有核苷酸的数量;IPtb表示由文献[6]中整体三联体-核苷酸倾向性度量公式(1)计算得到的三联体t和核苷酸b的相互作用倾向性值,它表示的是三联体t和核苷酸b的整体倾向性,而本文中式(1)计算的PRIPtb值表示的是一对蛋白质-RNA序列个体中三联体t和核苷酸b的相互作用倾向性.以下均用三联体-核苷酸的整体倾向性表示来自于文献[6]中的度量公式(1)计算得到的倾向性,即IPtb;用三联体-核苷酸的个体倾向性表示由本文中的权重倾向性度量公式(1)计算得到的倾向性,即PRIPtb.
1.2 构建特征向量
为了预测一对蛋白质-RNA序列是否相互作用,利用氨基酸三联体-核苷酸的个体倾向性编译特征向量.首先,根据极性和侧链容积等性质,把20种氨基酸分成7类[5],依次是:{A,G,V},{I,L,F,P},{Y,M,T,S},{H,N,Q,W},{R,K},{D,E},{C}.在文献[5]中,作者利用氨基酸三联体有效地预测蛋白质-蛋白质相互作用.本文中也同样使用三联体特征.20种氨基酸被分成7类,此时三联体共有7×7×7=343类,依次可以计算出三联体-核苷酸的组合个数为343×4=1372.给定一对蛋白质-RNA序列,构造如下特征向量:
1.2.1 个体氨基酸三联体-核苷酸倾向性
第一,利用整体倾向性度量公式[6]分别计算出所有32 000个三联体-核苷酸组合的相互作用倾向性值IPtb;
第二,基于32000个整体倾向性IPtb,计算每类三联体-核苷酸倾向性的均值,用来表示这类三联体-核苷酸的倾向性值,共有343×4=1372个倾向性;
第三,针对每一对蛋白质-RNA序列,利用权重倾向性度量公式(1)计算这对序列中每类三联体-核苷酸的个体倾向性,并以此作为特征向量.此时式(1)中的IPtb表示的是由第二步计算得到的每类三联体-核苷酸倾向性均值,Nt表示的是蛋白质序列中每类三联体的数量;
第四,考虑到组合特征的冗余性,从中选择具有较高倾向性的三联体-核苷酸组合,并以这些三联体-核苷酸组合为基础建立特征向量.
1.2.2 氨基酸三联体和核苷酸成分特征
第一,对于一个蛋白质序列,计算343类三联体的成分比率;
第二,对于一个RNA序列,计算4种核苷酸的成分比率.
1.3 样本数据
为了证明预测的有效性,本文主要针对两组不同种类的数据集进行预测:一组是来自NPInter数据库(http://www.bioinfo.org.cn/NPInter)的367对长链非编码RNA(简称ncRNA)和蛋白质相互作用的数据集PRI367,见表1;另一组是来自PRIDB[7]的非冗余数据集PRI369[2],见 表2.PRIDB是一个从PDB[8]里提取的一个综合的蛋白质和RNA复合物的数据库.

表1 RPI367计算结果
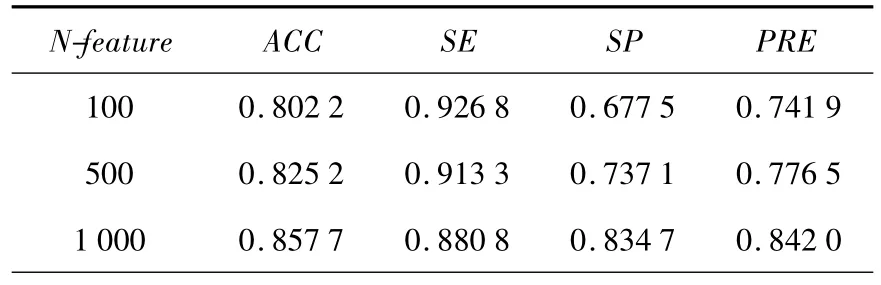
表2 RPI369计算结果
负样本数据的选取我们采纳大多数文献中的方法,即随机选取与正样本数据相同数量的蛋白质和RNA序列对作为负样本数据集,但是前提是排除那些已知有相互作用的蛋白质-RNA对.
1.4 预测模型和算法
支持向量机(SVM)是Vapnik等人提出的一类新型机器学习方法,是基于统计学习理论、根据结构风险最小化原理而推导出来的.由于SVM出色的学习能力,普遍应用于生物信息学研究中,很多生物信息学中的分类问题都是利用SVM进行分类的.本文也采用SVM对蛋白质-RNA是否有相互作用进行分类预测.
这里简单地介绍一下支持向量分类机的模型:
对于一个给定的训练集
T={(x1,c1),(x2,c2),…,(xl,cl)}
其中,xi=(xi1,xi2,…,xin)T∈Rn是输入(input),表示第i个输入样本的n个特征;ci∈{-1,+1}是输出(output),表示第i个样本所属的分类.引入从输入空间到Hilbert空间的映射φ:Rn→H.支持向量机就是为了寻找一个Hilbert空间的超平面(ω·φ(x))+b=0,使得在最大间隔的基础上将样本尽可能的分开.通过使用核函数替代样本在Hilbert空间中的内积,来判别样本所属类别.考虑到RBF核函数优于其他核函数,本文使用RBF核函数.
本文利用公开软件LibSVM(version 3.18)训练SVM中的C-SVC,其性能依赖于参数的选择,所需选择的参数为:C和gamma.其中C是惩罚参数,是对错分点的惩罚;gamma是RBF核函数中的参数,它决定向量机的推广能力.
2 计算结果及讨论
在计算中,参数 C=200和gamma=0.1.使用10折交叉验证程序评价我们的预测算法,预测结果的有效性主要考虑了以下几个指标:
ACC=(TP+TN)/(TP+FP+TN+FN)
SE=TP/(TP+FN)
SP=TN/(TN+FP)
PRE=TP/(TP+FP)
其中,TP表示真的正样本(true positives);TN表示真的负样本(true negatives);FP表示假的正样本(false positives)和FN表示假的负样本(false negatives);ACC(正确率),SE(灵敏度),SP(特指度)和PRE(精度).
针对两组数据集PRI367和PRI369,分别取100,500,1000个三联体-核苷酸组合特征建立特征向量进行计算,结果见表1和表2.其中N-feature表示所选取的三联体-核苷酸倾向性的个数.由计算结果可以看出,随着所选特征的增加,RPI369的正确率有一定的提高,而RPI367的正确率反而降低了.当特征个数增加到1 000时,正确率没有太大的变化.而在目前仅仅基于序列预测蛋白质 -RNA相互作用的工作[3]中,对RPI369数据集使用两种分类方法(RF和SVM)实施10折交叉验证,正确率分别为76.2%和72.8%.在以前的研究工作中,基于 Naïve Bayesian的分类方法对RPI367和RPI369进行10折交叉验证,正确率仅仅达到77.6%和75.0%.通过比较,可以看出本文的计算结果更好一些.
氨基酸三联体和核苷酸的相互作用倾向性被用于预测RNA结合位点得到了很好的预测结果,于是我们试图把它运用到蛋白质-RNA相互作用的预测中.考虑到每对蛋白质-RNA序列中三联体-核苷酸的倾向性的差别,重新定义了一个权重倾向性度量,然后利用此度量计算每类三联体-核苷酸的倾向性.计算结果证实了本文所选特征的有效性,同时也说明了三联体-核苷酸的相互作用倾向性在蛋白质-RNA相互作用预测中起着不可忽视的重要作用.
[1]PANCALDI V,BAHLER J.In silico characterization and prediction of global protein RNA interactions in yeast[J].Nucleic Acids Res.,2011,39:5826-5836.
[2]BELLUCCI M,AGOSTINI F,MASIN M,et al.Predicting protein associations with long noncoding RNAs[J].Nat.Methods,2011(8):444-445.
[3]MUPPIRALA U K,HONAVAR V G,DOBBS D.Predicting RNA-protein interactions using only sequence information[J].BMC Bioinformatics,2011,12:489.
[4]WANG Y,CHEN X W,LIU Z P,et al.De novo prediction of RNA-protein interactions from sequence information[J].Mol.BioSyst.,2013(9):133-142.
[5]SHEN J,ZHANG J,LUO X,et al.Predicting proteinprotein interactions based only on sequences information[J].Proc.Natl.Acad.Sci.U.S.A.,2007,104:4337-4341.
[6]CHOI S,HAN K.Prediction of RNA-binding amino acids from protein and RNA sequences.BMC Bioinformatics[J].2011,12(Suppl 13):7.
[7]LEWIS BA,WALIA R R ,TERRIBILINI M ,et al.PRIDB:a Protein-RNA Interface Database[J].Nucleic Acids Res.,2011,39:80-82.
[8]BERMAN H M ,WESTBROOK J,FENG Z,et al.The Protein Data Bank[J].Nucleic Acids Res.,2000,28:235-242.
猜你喜欢
九江职业技术学院学报(2022年1期)2022-12-02 09:46:54
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
保定学院学报(2022年2期)2022-04-07 02:26:50
有色金属(矿山部分)(2021年4期)2021-08-30 06:10:42
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
许昌学院学报(2018年4期)2018-05-02 12:27:37
中华建设(2017年1期)2017-06-07 02:56:14
新闻研究导刊(2015年17期)2015-12-25 12:36:42
语言与翻译(2015年4期)2015-07-18 11:07:43
