
肌原纤维肌病的临床与病理特征(附1例报告)☆
2015-02-06 09:04陈裕庆刘文文高美钦吴敏霞张文敏
中国神经精神疾病杂志 2015年4期
陈裕庆刘文文高美钦吴敏霞张文敏
·论 著·
肌原纤维肌病的临床与病理特征(附1例报告)☆
陈裕庆*刘文文*高美钦*吴敏霞*张文敏*
目的研究肌原纤维肌病(myofibrillar myopathy,MFMs)的临床与病理学特征。方法回顾性分析1例肌原纤维肌病患者的临床与病理学资料。结果本例为散发病例,青年女性患者,病程较短,主要表现为四肢近端肌无力伴轻度肌萎缩,骨骼肌病理活检示病变肌纤维内出现空泡,内有紫红色颗粒状或团块状物质沉积,结蛋白、dysferlin及a1-抗胰蛋白酶抗体免疫组化染色阳性,电镜示部分肌原纤维破坏,Z线消失,可见肌原纤维降解产物堆积;对症治疗后患者症状缓解,随访1年未再发肌无力等临床表现。结论肌原纤维肌病临床表现缺乏特异性,病情进展缓慢,确诊有赖于骨骼肌病理活检,光镜见肌纤维内结蛋白等多种蛋白质异常沉积,电镜见肌原纤维破坏、Z线消失。
肌原纤维肌病结蛋白临床特点病理特征
肌原纤维肌病(myofibrillar myopathy,MFMs)是一类罕见的神经肌肉疾病,成人多见,临床表现缺乏特异性,主要表现为慢性进行性四肢肌无力,可伴有心肌受累和周围神经病,确诊有赖于骨骼肌病理活检,主要表现为肌纤维肌原纤维破坏崩解伴Z线及其相关物质或包涵体的异常聚集。该病目前国内研究较少,仅见1个家系和3个散发病例报告。现报告1例经骨骼肌病理活检确诊的肌原纤维肌病,并分析其临床与病理学特征,以提高对该病的认识。
1 资料与方法
1.1 资料患者,女,20岁,因“全身肌肉酸痛伴四肢乏力2周”入院。入院前2周无明显诱因出现全身肌肉酸痛不适,以四肢近端为甚,活动后加重,休息后改善,伴四肢乏力,双肩上举、下蹲费力,无胸闷、气促,无恶心、呕吐,无腹痛、腹胀、腹泻。患者既往体健,否认家族中有类似症状成员。体格检查:四肢活动尚自如,无关节红肿、畸形,肌肉轻度萎缩,近端肌力5-级,远端肌力5级,肌张力正常对称,无肌肉痉挛、疼痛及麻木,生理反射正常对称,病理征未引出;心脏、双肺及腹部检查无异常。辅助检查:乳酸脱氢酶210 U/L,a-羟丁酸脱氢酶178 U/L,肌酸激酶84 U/L,肌酸激酶同工酶13 U/L,血沉7 mm/h,C-反应蛋白<0.824 mg/L,自身抗体全套阴性,TORCH全套阴性;肌电图示肌源性损害;心电图示正常心电图。
1.2 方法
1.2.1 骨骼肌活检 取左侧肱二头肌行开放性肌肉活检,标本分为两份:一份经液氮冷却的异戊烷速冻后,制作冰冻切片;另一份标本经2.5%戊二醛和1%锇酸双重固定、梯度丙酮脱水、环氧树脂浸透和包埋后,制作超薄切片。
1.2.2 普通染色 取冰冻切片,行苏木精-伊红(he⁃matoxylin and eosin,HE)染色,光学显微镜下观察结果并拍照。
1.2.3 特殊染色取冰冻切片,行改良Gomori三色(modified gomori trichrome,MGT)、还原型辅酶Ⅰ四唑氮还原酶(nicotinamide adenine dinucleotide tetra-zolium reductase,NADH-TR)、腺苷三磷酸酶(adenosine triphosphatase,ATPase)、琥珀酸脱氢酶
(succinate dehydrogenase,SDH)、细胞色素氧化酶(cytochrome oxidase,COX)、酸性磷酸酶(acid phos⁃phatase,ACP)、过碘酸-雪夫(periodic acid schiffs, PAS)、油红O(Oil Red O)及刚果红(congo red)染色,光学显微镜下观察结果并拍照。
1.2.4 免疫组织化学染色 取冰冻切片,4℃下冷丙酮固定10 min后,行抗结蛋白(desmin,即用型)、dysferlin(浓度1:20)、α1-抗胰蛋白酶(α1-antitryp⁃sin,即用型)、肌动蛋白(actin,即用型)、肌球蛋白(myosin,即用型)、泛素(ubiquitin,即用型)、细胞角蛋白(cytokeratin,即用型)、抗肌萎缩蛋白(dys⁃trophin,浓度1:40)及sarcoglycan(浓度1:100)抗体免疫组化染色(EliVision二步法),二氨基联苯胺(DAB)显色,光学显微镜下观察结果并拍照。
1.2.5 透射电子显微镜观察 取超薄切片,行铅-铀双重染色,透射电镜下观察并拍照。
2 结果
2.1 HE染色部分肌纤维轻度萎缩,肌质内可见大小不等、形态不规则的空泡,内有紫红色颗粒状或团块状物质沉积(图1),未见肌纤维坏死及吞噬反应,核内移无增多,间质未见炎症细胞浸润。
2.2 组织化学染色MGT染色见部分肌纤维内不规则斑块状蓝紫色物质沉积(图2);NADH-TR染色见部分肌纤维内氧化酶活性增高,呈深染的斑块状(图3);ATP酶染色见部分肌纤维内局灶酶活性缺失,两型肌均有受累(图4);SDH、COX及ACP染色未见异常;PAS、油红O及刚果红染色阴性。
2.3 免疫组织化学染色抗desmin染色见部分肌纤维内棕黄色颗粒状物质沉积,呈阳性反应(图5)。此外,抗dysferlin和抗α1-antitrypsin染色亦呈阳性反应(图6、7)。抗actin、myosin、ubiquitin、cy⁃tokeratin、dystrophin及sarcoglycan染色阴性。
2.4 透射电镜观察部分肌原纤维排列紊乱或破坏缺失,Z线消失,可见肌原纤维降解产物堆积(图8)。
3 讨论
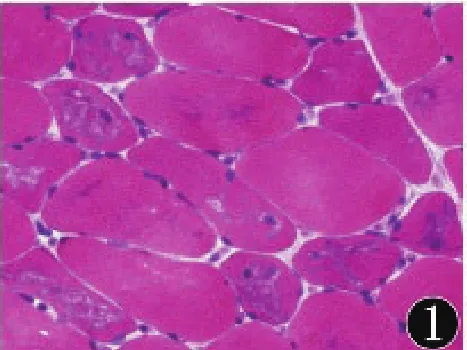
图1 肌纤维内出现空泡,内有紫红色物质沉积(HE染色,400×)
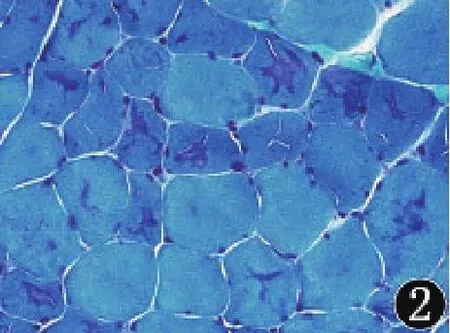
图2 肌纤维内不规则斑块状蓝紫色物质沉积(MGT染色,400×)

图3 肌纤维内可见氧化酶活性增高的深染区(NADH-TR染色,400×)

图4 肌纤维内局灶ATP酶活性缺失,两型肌均受累(ATP酶染色,pH10.4,400×)

图5 肌纤维内出现结蛋白异常沉积(抗desmin染色,400×)
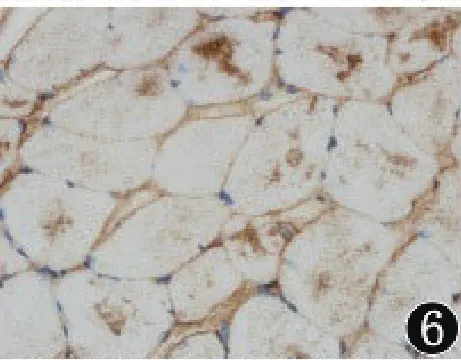
图6 肌纤维内出现dysferlin异常沉积(抗dysferlin染色,400×)

图7 肌纤维内出现α1-抗胰蛋白酶异常沉积(抗α1-antitrypsin染色,400×)

图8 肌原纤维排列紊乱,Z线消失,可见肌原纤维降解产物堆积(铅-铀双重染色,8000×)
MFMs是一类具有临床和遗传异质性的神经肌肉疾病[1],病因未明,发病年龄较广泛,但以成人多见,常为散发发病,家族遗传性病例多呈常染色体显性遗传,少数为常染色体隐性遗传。临床上表现为缓慢进展的肢体无力,可累及近端,但以远端受累更常见,有些伴有肌肉萎缩、僵硬、痉挛、疼痛或感觉异常等症状,部分患者出现心肌、周围神经或胃肠道受累表现,肌酶多为正常或轻度增高,
肌电图提示肌源性损害或合并神经源性损害[2],可见纤颤、正相电位、重复电位发放或肌强直样放电。总体来说,MFMs临床表现缺乏特异性,确诊有赖于骨骼肌病理活检。
MFMs共同的病理学特征是病变肌纤维内出现结蛋白的异常沉积,故早期被命名为“结蛋白肌病”或“结蛋白相关性肌病”。后来研究发现,肌纤维内沉积物中还可以存在其它蛋白质的聚集,这些蛋白质均与肌原纤维Z线相关,因此将这类疾病称为“肌原纤维肌病”。形态学上典型病变表现为部分肌纤维内可出现肌膜下和/或肌质内红染或蓝染的物质聚集、胞浆体、镶边空泡、非镶边空泡、擦除肌纤维、轴空样纤维[3-4];此外,多数病例肌纤维大小不等,可见萎缩和肥大肌纤维,有些病例还可见撕裂纤维、核内移增多,肌纤维坏死及炎症反应一般不明显。MGT染色显示肌纤维内深蓝色或暗绿色的异常结构,少数可出现破碎红纤维;NADH-TR染色可见氧化酶活性增高的深染物质沉积或氧化酶活性显著降低的轴空样纤维;部分病例COX染色可见较大范围的酶活性缺失区,称之为“擦除肌纤维”。免疫组化染色可检测到多种蛋白质异常沉积,如结蛋白、αβ-晶体蛋白、syncoi⁃lin、泛素、肌收缩蛋白、细丝蛋白C、窖蛋白-3、抗肌萎缩蛋白、β-淀粉样前体蛋白、细丝状肌动蛋白、凝溶胶蛋白、热休克蛋白、神经细胞黏附分子、磷酸化Tau蛋白、Tar DNA结合蛋白、p62蛋白、朊病毒蛋白及α1-抗胰蛋白酶等,但诊断MFMs并不需要所有蛋白质染色均阳性,其中最有意义的是结蛋白、抗肌萎缩蛋白、肌收缩蛋白及αβ-晶状体蛋白。电镜检查显示不同程度的肌原纤维破坏,Z线水纹状改变,Z线物质堆积、颗粒状或细丝状物质聚集及各种包涵体、髓样小体、自噬碎片等结构[5]。
目前已发现7个基因异常与MFMs发病相关[6],包括结蛋白(desmin)、αβ-晶体蛋白(αβ-crystal⁃lin)、肌收缩蛋白(myotilin)、Z线选择性剪接PDZ蛋白(ZASP)、细丝蛋白C(FLNC)、Bcl-2相关抗凋亡蛋白3(BAG3)及FHL1蛋白,但大量病例的缺陷基因尚未明确。除了一般的临床表现,不同基因缺陷引起的MFMs临床和病理特征尚有不同。结蛋白病心肌受累常见,可发生在肌无力之前、与肌无力同时出现或随病情的进展出现[7],表现为心律失常、扩张型心肌病或肥厚型心肌病;白内障可作为αβ-晶体蛋白病区别于其它MFMs的临床特征[8],此外,还可出现面肌无力、吞咽困难及构音障碍;肌收缩蛋白病、ZASP病及FLNC病大多发病较晚(患者年龄超过40岁);肌收缩蛋白病的临床表现类似肢带型肌营养不良1A,肌无力累及近端肌和远端肌,部分伴有关节挛缩及呼吸功能不全;FLNC病可出现关节挛缩和手部肌肉萎缩;BAG3病亦可出现关节挛缩,患者若儿童期发病,则病情进展快,且心肌病、脊柱强直和侧弯是其临床特征,当病变累及周围神经时,肌电图可检测到肌强直电位;FHL1病可表现为不对称性肌无力,儿童患者病情进展迅速,可伴发脊柱强直和侧弯。病理形态学上,结蛋白病可出现PAS阳性空泡[9],球形体的出现高度提示肌收缩蛋白病[10],还原体常出现在FHL1病[11],擦除肌纤维更多出现在结蛋白病和αβ-晶体蛋白病,而镶边空泡和非镶边空泡较为突出地出现在肌收缩蛋白病和ZASP病[4]。尽管已发现上述基因异常与MFMs发病相关,但其病因仍不明确,大量病例的缺陷基因尚未确定,骨骼肌病理活检仍是确诊MFMs的主要手段,病变肌纤维内结蛋白等多种蛋白质异常沉积是确诊的主要依据[12-13]。
本例患者为青年女性,四肢近端无力伴肌肉轻度萎缩,无心肌及周围神经等受累表现,未见特异性临床表现。骨骼肌病理活检HE和MGT染色均可见病变肌纤维内颗粒状或团块状物质沉积;NADH-TR染色示肌纤维内异常沉积物氧化酶活性增高;抗desmin、dysferlin及α1-antitrypsin免疫组织化学染色阳性,证实肌纤维内沉积物包含结蛋白、dysferlin及α1-抗胰蛋白酶,而在目前国内报告的病例中,对肌纤维内沉积物种类的检测较少,仅检测到结蛋白的异常沉积;透射电镜示部分肌原纤维破坏,Z线消失,肌原纤维降解产物堆积;以上病变符合MFMs的典型病理学特征,可确
诊为MFMs。临床予以对症治疗,患者症状缓解,出院后随访一年,未再发肌无力等临床表现,提示本病进展缓慢。
[1]Selcen D,Ohno K,Engel AG.Myofibrillar myopathy:clinical, morphological and genetic studies in 63 patients[J].Brain, 2004,127(2):439-451.
[2]Bergman JE,Veenstra-Knol HE,van Essen AJ,et al.Two relat⁃ed Dutch families with a clinically variable presentation of car⁃dioskeletal myopathy caused by a novel S13F mutation in the desmin gene[J].Eur J Med Genet,2007,50(5):355-366.
[3]Goebel HH,Fardeau M,Olivé M,et al.156th ENMC Interna⁃tional Workshop:desmin and protein aggregate myopathies, 9-11 November 2007,Naarden,The Netherlands[J].Neuromus⁃cul Disord,2008,18(7):583-592.
[4]Claeys KG,van der Ven PF,Behin A,et al.Differential involve⁃ment of sarcomeric proteins in myofibrillar myopathies:a mor⁃phological and immunohistochemical study[J].Acta Neuro⁃pathol,2009,117(3):293-307.
[5]Victor Dubowitz,Caroline A.Sewry,Anders Oldfors.Muscle Bi⁃opsy:A Practical Approach[M].Saunders,2013.
[6]Schröder R,Schoser B.Myofibrillar myopathies:a clinical and myopathological guide[J].Brain Pathol,2009,19(3):483-492.
[7]Kostera-Pruszczyk A,Pruszczyk P,Kamińska A,et al.Diversity of cardiomyopathy phenotypes caused by mutations in desmin [J].Int J Cardiol,2009,131(1):146-147.
[8]Selcen D,Engel AG.Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations[J].Ann Neurol, 2003,54(6):804-810.
[9]Clemen CS,Fischer D,Reimann J,et al.How much mutant pro⁃tein is needed to cause a protein aggregate myopathy in vivo? Lessons from an exceptional desminopathy[J].Hum Mutat, 2009,30(3):490-499.
[10]Foroud T,Pankratz N,Batchman AP,et al.A mutation in myoti⁃lin causes spheroid body myopathy[J].Neurology,2005,65(12): 1936-1940.
[11]Shalaby S,Hayashi YK,Goto K,et al.Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene(FHL1)[J].Neuromuscul Disord,2008,18(12):959-961.
[12]Olivé M,Kley RA,Goldfarb LG.Myofibrillar myopathies:new developments[J].Curr Opin Neurol,2013,26(5):527-535.
[13]Claeys KG,Fardeau M.Myofibrillar myopathies[J].Handb Clin Neurol,2013,113:1337-1342.
R746
A
2014-07-18)
(责任编辑:李立)
10.3936/j.issn.1002-0152.2015.04.011
☆ 福建省省属高校项目(编号:JK2012017),福建省教育厅科技项目(编号:JA13144)
* 福建医科大学基础医学院病理学系(福州350004)
猜你喜欢
食品工业科技(2022年17期)2022-08-27
中国医院院长(2021年21期)2022-01-21
今日农业(2021年5期)2021-11-27
中华养生保健(2021年18期)2021-02-13
医学信息(2020年12期)2020-07-27
世界科学技术-中医药现代化(2020年2期)2020-07-25
食品科学(2020年11期)2020-07-13
中国现代神经疾病杂志(2020年1期)2020-01-08
中国中医急症(2019年10期)2019-05-21
中国社区医师(2019年4期)2019-01-18
