
星形胶质细胞对天冬氨酸特异性半胱氨酸蛋白酶介导β淀粉样蛋白早期突触毒性作用的影响
2015-01-20 21:36吕昂初宋一志张亚丽李彦方圆陆涛刘津平常丽荣武艳
中国卒中杂志 2015年3期
吕昂初,宋一志,张亚丽,李彦,方圆,陆涛,刘津平,常丽荣,武艳
血管性痴呆(vascular dementia,VaD)是由于缺血或出血性脑血管疾病以及脑缺血、缺氧引起的认知功能障碍。血脑屏障的损伤在VaD的发生及发展中有着重要的作用。血脑屏障是存在于脑组织和血液之间的一个复杂的系统,控制血脑两侧的物质运输,从而保证中枢神经系统内环境的相对稳定[1]。星形胶质细胞(astrocyte,AS)是中枢神经系统的管家细胞,AS不但参与代谢物质的转移,释放神经营养因子,信号转导等,它还参与血脑屏障的形成[2]。已有研究结果表明VaD的病理机制中涉及AS的改变和炎性因子的分泌,并且毒性物质β-淀粉样蛋白(β-amyloid,Aβ)参与了VaD的发生发展[3-5]。本课题组前期研究已证实caspase参与介导了Aβ所诱导的神经元早期突触毒性作用[6]。但AS是否对caspase介导的Aβ毒性作用有影响尚不明确。本研究沿用以往研究选取Aβ作用1 h为早期突触毒性的时间点[6],应用免疫荧光方法,通过检测不同培养体系中[纯神经元体系(NE-S)及混合培养体系(MIX-S)],以特异性caspase-8/9抑制剂分别干预死亡受体/线粒体通路后对Aβ所诱导的突触后密度蛋白(postsynaptic density-95,PSD95)表达变化的影响,从而探讨AS对caspase介导Aβ早期突触毒性作用的影响,为深入研究VaD的发病机制提供理论基础。
1 对象和方法
1.1 研究对象 选用出生后4 d的Wistar清洁级大鼠(由北京华阜康生物科技公司提供),不分雌雄。
1.2 研究方法
1.2.1 建立海马细胞原代混合培养体系(MIX-S),将研究对象低温麻醉,于4℃解剖液中,在尸体解剖镜下分离海马,并机械切碎,加0.05%胰蛋白酶于37℃水浴摇床中震荡消化10 min,加入培养液,即含胰蛋白酶抑制剂和2%神经细胞生长添加剂(B27)的神经基础无血清培养基(Neurobasal-A)终止消化。电动移液器反复吹打使组织分散为单细胞,1000转/分离心使未打散的组织沉淀,0.22 μm细胞筛网过滤收集上清,重复上述步骤。所得上清离心,离心后弃上清,加37℃培养液吹打成单细胞悬液,配制不同浓度的牛血清白蛋白(bovine serum albumin,BSA)培养液进行梯度密度离心后,去除含细胞碎片的上清液,加2 ml培养液重新吹打成细胞悬液。细胞计数,75 cm2培养瓶内差速贴壁30 min,调整细胞密度至400×106/L,种于多聚右旋赖氨酸(Poly-DLysine,PDL)包被的小玻片上,30 min后加入含0.1 mg/ml卡那霉素、1 mg/ml谷氨酰胺替代物(glutamax)、10 ng/ml纤维原细胞基础生长因子(fibroblast growth factor-basic,bFGF)的Neurobasal-A培养液,放入37℃、5%CO2培养箱培养,每隔2 d半量换培养液1次,于第7天细胞状态良好时用于实验。
1.2.2 建立海马原代神经元纯化培养体系(NE-S) 前期种植细胞步骤同1.2.1,根据AS的生长曲线特点,于细胞开始种植18~24 h在培养液内加入5 μmol/L终浓度的阿糖胞苷,抑制AS的增殖,以后每隔2 d半量换培养液1次,于第7天细胞状态良好时用于实验。
1.2.3 分组处理 第7天用40 μmol/L的caspase-8抑制剂(Z-IETD-FMK)和caspase-9抑制剂(Z-LEHD-FMK)分别处理NE-S和MIX-S的caspase-8抑制剂组、caspase-9抑制剂组、caspase-8抑制剂预处理加Aβ组及caspase-9抑制剂预处理加Aβ组,于37℃、5%CO2培养箱孵育1 h;接着用10 μmol/L的Aβ25-35分别处理NE-S和MIX-S的Aβ处理组、caspase-8抑制剂预处理加Aβ组及caspase-9抑制剂预处理加Aβ组,于37℃、5%CO2培养箱孵育1 h。
1.2.4 免疫细胞化学染色 加药处理后的细胞用4%多聚甲醛固定10 min,磷酸盐缓冲溶液(phosphate buffered saline,PBS)洗涤后,加入含0.3%聚乙二醇辛基苯基醚(TritonX-100)的PBS穿透20 min,马血清封闭液(含5%正常马血清的PBST)封闭20 min,加入一抗PSD95(1∶200)常温孵育2 h后入4℃冰箱过夜。PBS洗涤后加入荧光二抗,避光常温孵育2 h,PBS洗涤,细胞核荧光染料(Hoechst 33342)复染细胞核,封片观察。
1.2.5 图像采集及统计学分析 随机挑选形态完整的阳性神经元,荧光显微镜100倍油镜下摄取细胞图片,以Photoshop图像分析软件定量分析,每个神经元选取1~2个树突,计数近胞体10 μm段树突上PSD95阳性表达量,共计数30个以上神经元。计数样本至少来自5次独立实验(n=5)。实验数据经SPSS 13.0软件统计分析,符合正态分布,以(表示,采用方差分析做整体差异比较,如有统计学意义,各组之间采用LSD-t检验进行多重比较,检验标准α=0.05,P<0.05差异有显著性。
2 结果
2.1 NE-S各组PSD95表达情况 在NE-S各组,整体差异比较有显著性(F=9.53,P<0.001)。进一步两两比较显示:caspase-8抑制剂组(P=0.95)、caspase-9抑制剂组(P=0.98)与对照组相比,PSD95的表达量无明显变化;Aβ处理组与对照组相比PSD95的表达量降低,有显著差异(P<0.001);caspase-8抑制剂预处理加Aβ组与Aβ处理组相比PSD95的表达量无明显变化(P=1.00),与对照组相比则降低,有显著差异(P<0.001);caspase-9抑制剂预处理加Aβ组与Aβ处理组相比PSD95表达量回升,有显著差异(P<0.001),与对照组相比则无明显变化(P=0.97)(图1)。

图1 NE-S各组PSD95表达的免疫荧光图及统计结果
2.2 MIX-S各组PSD95表达情况 在MIX-S组,整体差异比较有显著性(F=8.58,P<0.001)。进一步两两比较显示caspase-8抑制剂组(P=0.92)、caspase-9抑制剂组(P=0.84)与对照组相比,PSD95的表达量无明显变化;Aβ处理组与对照组相比PSD95的表达量降低,有显著差异(P<0.001);caspase-8 抑制剂预处理加Aβ组与Aβ处理组相比PSD95的表达量升高,有显著差异(P<0.001),与对照组相比则无明显变化(P=0.99);caspase-9抑制剂预处理加Aβ组与Aβ处理组相比PSD95表达量无明显变化(P=0.90),与对照组相比则降低,有显著差异(P<0.001)(图2)。
2.3 MIX-S与NE-S相比,各组PSD95表达情况 与NE-S相比,MIX-S对照组(P=0.98)、caspase-8抑制剂组(P=0.96)、caspase-9抑制剂组(P=0.84)和Aβ处理组(P=0.37)PSD95的表达量无显著变化;MIX-S caspase-8抑制剂预处理加Aβ组,PSD95的表达显著高于NE-S组(P<0.001),MIX-S caspase-9抑制剂预处理加Aβ组PSD95的表达量显著低于NE-S组(P<0.001)(图3)。
3 讨论
随着人口老龄化,老年痴呆的发病率与患病率逐年上升,给社会带来了极大的负担。VaD是老年痴呆的重要类型,发病率仅次于阿尔茨海默病(Alzheimer’s disease,AD),而VaD可与AD并存并加重AD病情,故对于VaD的研究迫在眉睫[3,7]。有研究者发现,AD患者多数伴有脑血管病变,脑血管疾病可以影响Aβ的积聚[8],Aβ亦可导致血管收缩、增加脑组织缺血易损性[9-10]。另有研究报道,伴随着Aβ浓度的变化,海马区神经元丢失可成为VaD发生的病理基础[5,11]。众所周知,caspase通路主要有内源性的caspase-9所介导的线粒体通路和外源性的caspase-8所介导的死亡受体通路[12-14]。近年来研究结果表明,在神经突触明显丢失之前即可观察到认知功能障碍,且突触功能的改变发生于VaD疾病进程早期,即神经元未发生大量凋亡时[15-18]。突触蛋白的表达变化可反映突触功能的改变,本课题组以往研究已证实,Aβ在尚未诱导显著神经元凋亡的早期毒性作用中可诱导神经元的PSD95表达减少,且caspase参与介导了Aβ所诱导的神经元早期突触毒性作用[6]。因此本实验沿用突触标志蛋白PSD95作为观察指标。

图2 MIX-S各组PSD95表达的免疫荧光图及统计结果
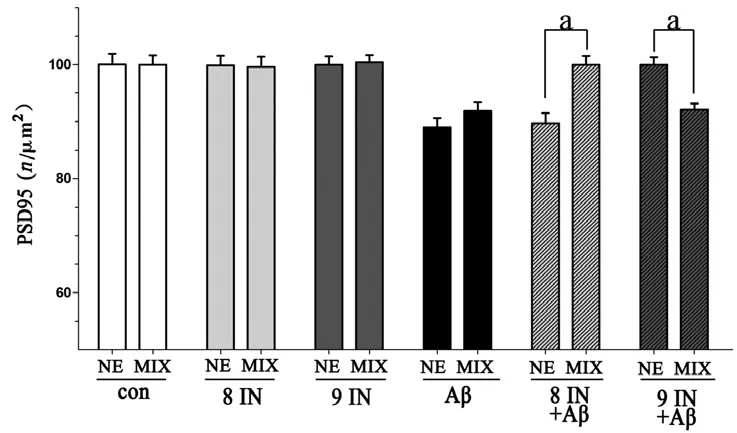
图3 NE-S与MIX-S各组PSD95表达量组间比较结果
本实验中,caspase-8/9的特异性抑制剂,可以分别有效地阻断死亡受体通路和线粒体通路。对细胞进行单纯的caspase-8/9抑制剂处理,与对照组相比PSD95的表达量并未发生变化,证实caspase-8/9抑制剂自身对突触没有毒性作用。在NE-S中,caspase-9抑制剂可显著缓解Aβ引起的PSD95减少,而caspase-8抑制剂并无显著影响。该结果提示在NE-S,Aβ所诱导的早期突触毒性中,caspase-9所介导的线粒体通路被激活,而caspase-8介导的死亡受体通路未参与其中,从而提示NE-S的海马神经元在Aβ早期突触毒性中发生了线粒体功能损伤;而在MIX-S则相反,提示AS的存在使得Aβ突触毒性作用机制发生改变,并证实AS对caspase介导Aβ早期突触毒性作用确有影响。但AS影响Aβ早期突触毒性作用的直接方式尚不明确。在中枢神经系统中,AS始终伴随神经元的整个发育过程,具有复杂而多样的功能。AS参与神经元物质代谢、突触可塑性调控和信号转导等功能,且与神经元损伤过程密切相关[19-20]。本课题组前期研究结果提示,Aβ作用24 h,AS参与保护Aβ诱导的海马神经元凋亡[21]。并且AS具有对Aβ酶切降解的能力,在Aβ清除中发挥重要作用[22-23]。近几年研究者发现,AS还可分泌可溶性的炎症因子,加速Aβ神经毒性作用[24-25];且在单纯AS系统中,Aβ刺激下的AS不仅失去保护功能,还可释放促凋亡物质[21]。在VaD患者脑中,TNF的表达量明显增加[3]。针对AS在Aβ诱导的神经毒性中作用不同的观点,结合两种不同的培养体系,本次研究结果揭示,由caspase-9所介导的线粒体通路在AS缺失的NE-S被激活,而在MIX-S无显著影响,提示AS可能是通过保护线粒体来干预Aβ早期突触毒性作用;有趣的是,由caspase-8所介导的死亡受体通路在AS缺失的NE-S未被激活,在AS存在的MIX-S被激活,提示AS的存在可能促使某种死亡受体配体分泌(如TNF),进而激活caspase-8所介导的死亡受体通路来参与Aβ早期突触毒性作用。
VaD进程中,Aβ浓度在脑内表达增加,而在血液中反而降低[5]。并且在AD中,Aβ毒性作用下,神经元未发生显著凋亡时,除突触功能发生改变外,线粒体功能也发生了变化,Aβ可通过与线粒体相结合导致神经元损伤[26-27]。结合本研究NE-S中线粒体通路被激活,这一机制也可能是VaD中神经元显著损伤的原因之一。本实验中AS作为一把双刃剑,扮演着不同角色。在VaD进程中,AS均有增生并发生形态改变以及TNF的表达量增加,且TNF的表达和AS的反应有相关性[3,24-25],提示VaD的发病机制非常复杂。AS在Aβ早期突触毒性作用的影响是否具有动态变化有待进一步研究,同时我们推测可能是AS释放了TNF进而激活死亡受体通路,亦待深入探索。AS在VaD病理进程中扮演着重要的角色,一系列的研究将为发现VaD的早期诊断、治疗的新靶点提供重要的理论依据。
1 温泽锋, 姜平, 童鑫康. 血脑屏障与血管性痴呆的研究进展[J]. 国外医学(老年医学分册), 2007, 28:211-216.
2 胡潇, 余涓. 星形胶质细胞在血脑屏障与神经元损伤中的作用[J]. 神经解剖学杂志, 2012, 28:330-334.
3 景玉红, 宋焱峰, 王子仁. 血管性痴呆大鼠星形胶质细胞增生与TNF-α的表达[J]. 中华神经医学杂志, 2004,3:418-421.
4 梁国聪, 石胜良, 毕桂南, 等. β淀粉样蛋白和tau蛋白在血管性痴呆发病机制中的作用[J]. 广西医科大学学报,2011, 28:661-664.
5 承欧梅, 钟世江, 宴勇, 等. 脑卒中和血管性痴呆β淀粉样蛋白测定及临床意义[J]. 重庆医学, 2004, 33:555-556.
6 Liu JP, Chang LR, Francesco R, et al. Amyloid-β induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors[J]. J Alzheimers Dis, 2010, 22:541-556.
7 邵福源, 黄流清, 薛红, 等. 血管性痴呆研究[J]. 中国临床神经科学, 2001, 9:92-95.
8 Sudduth TL, Weekman EM, Brothers HM, et al.β-amyloid deposition is shifted to the vasculature and memory impairment is exacerbated when hyperhomocysteinemia is induced in APP/PS1 transgenic mice[J]. Alzheimers Res Ther, 2014, 6:32-32.
9 周爱红, 王荫华. 阿尔茨海默病与血管性危险因素[J].中华老年心血管病杂志, 2005, 7:65-67.
10 赵万红, 罗超. 阿尔茨海默病与脑缺血的相互作用[J].中国老年学杂志, 2013, 33:5206-5209.
11 蔺心敬, 李吕力, 杨德刚, 等. 血管性痴呆大鼠海马神经元凋亡和蛋白表达实验研究[J]. 中国现代医学杂志,2007, 17:2326-2329.
12 Li P, Nijihawan D, Budihardjo I, et al. Cytochrome C dATP-dependent formation of Apaf-1/easpase-9 complex initiates an apoptoic protease cascade[J].Cell, 1997, 91:479-489.
13 Budihardjo I, Oliver H, Lutter M, et al. Biochemical of Bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis[J]. Annu Rev Cell Del Bio, 1999, 15:269-290.
14 Ashkenazi A, Dixit VM. Death receptors:signaling and modulation[J]. Science, 1998, 281:1305-1308.
15 Tiraboschi P, Hansen LA, Alford M, et al. The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease[J]. Neurology, 2000, 55:1278-1283.
16 Scheff SW, Price DA, Schmitt FA, et al. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment[J]. Neurobiol Aging, 2006,27:1372-1384.
17 Götz J, Ittner LM, Schonrock N, et al. An update on the toxicity of Abeta in Alzheimer's Disease[J].Neuropsychiatr Dis Treat, 2008, 4:1033-1042.
18 王菲. 血管性痴呆的分子机制及治疗学研究进展[J].中国神经精神疾病杂志, 2014, 40:317-320.
19 Allaman I, Bélanger M, Magistretti PJ. Astrocyteneuron metabolic relationships:for better and for worse[J]. Trends Neurosci, 2011, 34:76-87.
20 Takuma K, Baba A, Matsuda T. Astrocyte apoptosis:implications for neuroprotection[J]. Prog Neurobiol, 2004, 72:111-127.
21 宋一志, 高香红, 常丽荣, 等. 星形胶质细胞对β-淀粉样蛋白诱导神经元凋亡的影响[J]. 神经解剖学杂志,2013, 29:108-114.
22 Nielsen HM, Mulder SD, Beliön JA, et al. Astrocytic A beta 1-42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins[J]. Glia, 2010, 58:1235-1246.
23 Dorfman VB, Pasquini L, Riudavets M, et al.Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer's disease[J].Neurobiol Aging, 2010, 31:1743-1757.
24 Garwood CJ, Pooler AM, Atherton J, et al. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture[J]. Cell Death Dis, 2011, 167:1-9.
25 Qin H, Benveniste EN. ELISA methodology to quantify astrocyte production of cytokines/chemokines in vitro[J]. Methods Mol Biol, 2012,814:235-249.
26 Reddy PH, Manczak M, Mao P, et al. Amyloidbeta and mitochondria in aging and Alzheimer's disease:implications for synaptic damage and cognitive decline[J]. J Alzheimers Dis, 2010, 20:499-512.
27 Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease[J]. Science, 2004, 304:448-452.
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中学生物学(2021年8期)2021-11-02
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
山西农业大学学报(自然科学版)(2020年1期)2020-03-04
中国现代药物应用(2019年16期)2019-09-06
中医眼耳鼻喉杂志(2019年3期)2019-04-13
中国果业信息(2019年1期)2019-01-05
中成药(2017年6期)2017-06-13
