
毛细管气相色谱法测定注射用利培酮微球中有机溶剂残留量
2015-01-19 07:57马建芳朱瀛华任飞亮彭兴盛陈桂良
中国药业 2015年12期
马建芳,朱瀛华,任飞亮,刘 蔚,彭兴盛,陈桂良
(上海市食品药品检验所,上海 200131)
注射用利培酮微球(商品名恒德® )是由美国杨森公司研发生产的首个长效非典型性抗精神病药物,结合了长效剂型和非典型抗精神病药的优势,可显著改善精神分裂症引起的阳性症状和阴性症状[1-2],具有用药次数少、可持续释放药物、血药浓度平稳、不良反应发生率低、有效性和安全性高、患者依从性和耐受性好的优点[3],被广泛应用于临床。利培酮微球制备过程中使用的有机溶剂包括乙醇、乙酸乙酯和苯甲醇,在微球固化成型过程中,致密坚硬的高分子材料聚乳酸-羟基乙酸共聚物可阻碍有机溶剂的挥发,造成有机溶剂残留[4-5]。残留的有机溶剂会引起患者注射部位刺激或疼痛,影响药物在体内的释放,从而影响药效,因此应控制利培酮微球中的残留溶剂,以提高产品质量,保证用药安全。2010 年版《中国药典(二部)》中规定,乙醇和乙酸乙酯是第3 类溶剂,限度为0.5%,未对苯甲醇的限度作要求。本研究中建立了可同时测定注射用利培酮微球中的残留溶剂乙醇、乙酸乙酯和苯甲醇含量的毛细管气相色谱法,具有操作简单、灵敏度高、重复性好、专属性强、快速准确的特点,可作为利培酮微球中有机溶剂残留量检测的质量控制方法。
1 仪器与试药
6890A 型气相色谱仪(美国Agilent 公司)。注射用利培酮微球(美国杨森公司,批号为DJZSW00,DKZSG00,DKZSS00);乙醇、乙酸乙酯、乙酸丙酯、苯甲醇、苯乙醇和N,N-二甲基甲酰胺(DMF)均为色谱纯。
2 方法与结果
2.1 色谱条件及系统适用性试验
色谱柱:DB-624 型毛细管柱(94 %二甲基聚硅氧烷和6 %氰丙基苯聚硅氧烷,320 μm×30 m,1.8 μm);氢火焰离子化检测器(FID):检测器温度为250 ℃,进样口温度为220 ℃,氦气为载气,采用恒流模式,流速为1.0 mL/min,氢气流速为35 mL/min,空气流速为350 mL /min,尾吹气流速为25 mL /min;进样量:3 μL;分流比:10 ∶1;分析方法:程序升温,初始柱温130 ℃,平衡5 min,以70 ℃/min 的速度升温至240 ℃,平衡10 min。取对照品溶液依法进样测定,乙醇、乙酸乙酯、乙酸丙酯、苯甲醇和苯乙醇的理论板数均大于5 000,各有机溶剂色谱峰完全分离,分离度均大于2.5,色谱图见图1。
2.2 溶液制备
取DMF 适量,置1 000 mL 容量瓶中,加乙酸丙酯25 μL 和苯乙醇150 μL,用DMF 稀释至刻度,摇匀,作为内标溶液,其中乙醇和乙酸乙酯以乙酸丙酯为内标,苯甲醇以苯乙醇为内标。取内标溶液适量,置100 mL 容量瓶中,精密称取乙醇155.49 mg、乙酸乙酯50.49 mg 和苯甲醇177.54 mg,置100 mL 容量瓶中,用内标溶液稀释至刻度,摇匀,作为对照品贮备液。精密量取对照品贮备液5 mL,置50 mL 容量瓶中,用内标溶液稀释至刻度,摇匀,制成乙醇155.49 μg/mL、乙酸乙酯50.49 μg/mL 和苯甲醇177.54 μg/mL的溶液,作为对照品溶液。精密称取样品内容物100 mg,置10 mL容量瓶中,加内标溶液约7 mL,超声10 ~15 min,使微球溶解,放冷至室温,用内标溶液稀释至刻度,摇匀,作为供试品溶液。
2.3 方法学考察
专属性试验:取上述5 种溶剂适量,分别置10 mL 容量瓶中,加入DMF 稀释至刻度,摇匀,作为5 种溶剂的定位溶液。将上述定位溶液分别注入气相色谱仪,按拟订色谱条件依法进样测定,结果出峰顺序依次为乙醇、乙酸乙酯、乙酸丙酯、苯甲醇和苯乙醇,定位溶液中各组分的保留时间与对照品溶液中各组分保留时间一致,表明专属性较强。
线性关系考察:分别精密量取对照品贮备液0.25,0.50,1.00,2.00,2.50,5.00,6.00,7.50 mL,各置50 mL 容量瓶中,加内标溶液稀释至刻度,摇匀,即得系列质量浓度对照品溶液。按拟订色谱条件依法进样测定,记录色谱图,以各个残留溶剂的质量浓度(C)为横坐标、对应主峰面积与内标物峰面积的比值(Y)为纵坐标(乙醇和乙酸乙酯以乙酸丙酯为内标,苯甲醇以苯乙醇为内标)进行线性回归,得到回归方程和相关系数,详见表1。
精密度试验:取2.2 项下对照品溶液,重复进样6 次。计算得乙醇、乙酸乙酯和苯甲醇峰面积与对应内标物峰面积比值的RSD分别为2.63%,1.91%,0.94% ( n=6),表明仪器精密度良好。
加样回收试验:分别精密量取对照品贮备液2.50,5.00,7.50 mL,各置已精密称取注射用利培酮微球内容物500 mg 的50 mL 容量瓶中,各3 份,加内标溶液约35 mL,超声10 ~15 min,使微球溶解,放冷至室温,用内标溶液稀释至刻度,摇匀,作为低、中、高浓度供试品溶液。另精密称取本品500 mg,置50 mL 容量瓶中,加内标溶液约35 mL,超声10 ~15 min,使微球溶解,放冷至室温,用内标溶液稀释至刻度,摇匀,作为空白对照溶液。按拟订色谱条件依法进样测定,计算回收率。结果见表2。
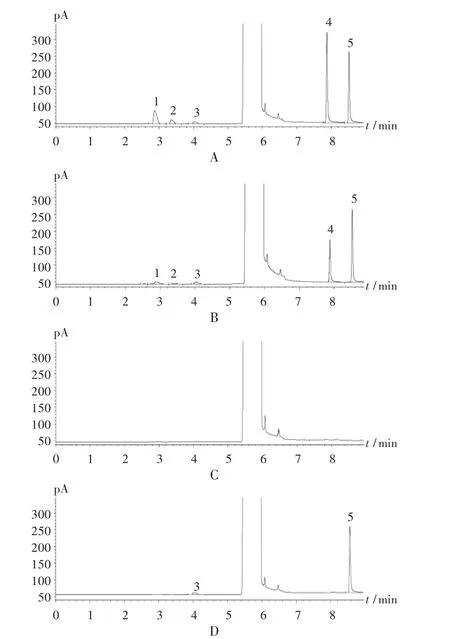
图1 气相色谱图
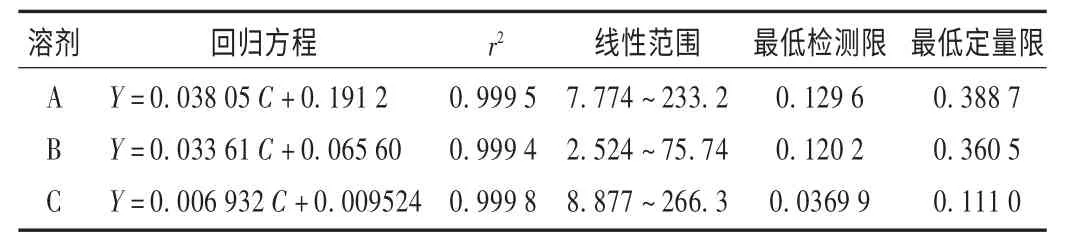
表1 线性关系考察结果及回归方程(μg/mL)
稳定性试验:按2.2 项下方法制备供试品溶液,取同一供试品溶液,按拟订色谱条件分别于0,2,6,12,24,36,48 h 时依法进样测定。结果乙醇、乙酸乙酯和苯甲醇峰面积与对应内标物峰面积比值的RSD 分别为3.43%,1.52%,1.02%(n=7),表明供试品溶液在48 h 内稳定。

表2 乙醇、乙酸乙酯和苯甲醇加样回收试验结果(n=9)
2.4 样品含量测定
按2.2 项下方法制备对照品溶液和供试品溶液,按拟订色谱条件分别依法进样测定,以内标法计算各有机溶剂残留量。结果见表3。

表3 样品含量测定结果(%)
3 讨论
进样方式和溶剂选择:微球的固化成型可能会影响制备过程中所用有机溶剂的挥发,残留的有机溶剂可影响药物稳定性,且增加毒副作用,故残留的有机溶剂量不应超过2010 年版《中国药典(二部)》的限量规定[6]。残留溶剂测定方法中,常用的进样方式主要有顶空进样和直接进样。本试验的预试验结果表明,采取直接进样法时,乙醇、乙酸乙酯、乙酸丙酯、苯甲醇和苯乙醇的分离度较好,各组分间不相互干扰,灵敏度高,相较顶空进样,可避免顶空没有完全平衡和顶空进样等带来的测定误差,且顶空进样装置容易引起乙醇和苯甲醇的残留,故本方法采用溶液直接进样。直接进样时,样品应完全溶解于溶剂中,经试验,利培酮微球中的主要辅料高分子材料聚乳酸-羟基乙酸共聚物可完全溶解于DMF 中,DMF 的沸点较高,很少干扰其他残留溶剂的检查。结果表明,采用直接进样法,利培酮微球中的辅料及溶剂DMF 对被测溶剂及内标物均无干扰,各组分峰之间的分离度均大于2.5。
色谱柱选择:预试验中比较了不同极性的毛细管色谱柱的分离效果,由于5 个溶剂的极性有较大区别,HP-1 和HP-5 等非极性色谱柱和DB-WAX 等极性色谱柱的分离效果均不理想,而中等极性色谱柱DB-624 对各溶剂峰的分离效果最优,且待测组分灵敏度高。在极性差异的基础上,考虑到各个溶剂的沸点差异,为实现各色谱峰的有效分离并尽量缩短分析时间,采用了程序升温的方法。比较了不同的起始柱温(120,130,140,150,160,170,180 ℃)和不同的升温速率(40,50,60,70,80 ℃/min)条件下各溶剂峰的分离效果。结果,130 ℃保持5 min 可使低沸点的乙醇、乙酸乙酯和乙酸丙酯尽早出峰并有效分离,然后以70 ℃/min的速度升温至240 ℃保持10 min 的程序升温条件,可显著缩短高沸点溶剂苯甲醇和苯乙醇的出峰时间,基线漂移范围小,可显著改善峰形,各溶剂峰均得到有效分离,避免微球辅料在毛细管柱中残留的现象出现。
定量方式选择:内标法可避免样品处理或进样体积误差对结果的影响,在测定挥发性残留溶剂时定量较准确。内标物应不干扰被测物的出峰,响应较好,为保证内标物的定量准确,内标物的保留时间应尽可能与被分析物质接近。苯甲醇的沸点显著高于乙醇和乙酸乙酯,为保证定量准确,分别采用乙酸丙酯和苯乙醇作为乙醇、乙酸乙酯和苯甲醇的内标,定量准确。结果表明,各残留溶剂的线性关系良好,精密度和准确度高,回收率符合要求。
[1] Park EJ,Amatya S,Kim MS,et al.Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia[J] .Arch Pharm Res,2013,36(6):651 -659.
[2] Nielsen J,Jensen SO,Friis RB,et al. Comparative effectiveness of risperidone long-acting injectable vs first-generation antipsychotic long-acting injectables in schizophrenia:results from a nationwide,retrospective inception cohort study[J].Schizophr Bull,2015,41(3):627 -636.
[3] 王丽莉. 第一个长效非典型抗精神病药—— 注射用利培酮微球[J].中国新药杂志,2010,19(22):2 066 -2 068.
[4] 左志辉,王 卫. 利培酮中有机溶剂残留的顶空毛细管气相色谱法测定[J]. 中国医药工业杂志,2013,44(5):496 -498.
[5] 张 华,王 翔,王学清,等. 注射用利培酮微球的制备和体外性质评价[J]. 中国新药杂志,2013,22(20):2 423 -2 426.
[6] 国家药典委员会. 中华人民共和国药典(二部)[M]. 北京:中国医药科技出版社,2010:附录ⅧP 61 -65.
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
昆明医科大学学报(2021年8期)2021-08-13
潍坊学院学报(2020年6期)2020-11-22
中成药(2017年8期)2017-11-22
中成药(2017年6期)2017-06-13
中成药(2017年3期)2017-05-17
浙江化工(2017年4期)2017-05-11
中国实用医药(2016年29期)2016-12-26
糖尿病新世界(2016年16期)2016-12-09
中国实用医药(2016年27期)2016-11-30
