
In silico modification of oseltamivir as neuraminidase inhibitor of influenza A virus subtype H1N1
2015-01-09 11:51UsmanSumoFriendTambunanRizkyArchintyaRachmaniaArliAdityaParikesit
Usman Sumo Friend Tambunan,Rizky Archintya Rachmania,Arli Aditya Parikesit
Bioinformatics Research Group,Department of Chemistry,Faculty of Mathematics and Natural Science,University of Indonesia,Depok Campus,Depok 16424,Indonesia.
In silico modification of oseltamivir as neuraminidase inhibitor of influenza A virus subtype H1N1
Usman Sumo Friend Tambunan✉,Rizky Archintya Rachmania,Arli Aditya Parikesit
Bioinformatics Research Group,Department of Chemistry,Faculty of Mathematics and Natural Science,University of Indonesia,Depok Campus,Depok 16424,Indonesia.
This research focused on the modification of the functionalgroups of oseltamivir as neuraminidase inhibitor against influenza A virus subtype H1N1.Interactions of three of the bestligands were evaluated in the hydrated state using molecular dynamics simulation at two different temperatures.The docking result showed that AD3BF2D ligand(N-[(1S,6R)-5-amino-5-{[(2R,3S,4S)-3,4-dihydroxy-4-(hydroxymethyl)tetrahydrofuran-2-yl]oxy}-4-formylcyclohex-3-en-1-yl]acetamide-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylate)had better binding energy values than standard oseltamivir.AD3BF2D had several interactions,including hydrogen bonds,with the residues in the catalytic site of neuraminidase as identified by molecular dynamics simulation.The results showed that AD3BF2D ligand can be used as a good candidate for neuraminidase inhibitor to cope with influenza A virus subtype H1N1.
influenza A virus subtype(H1N1),influenza,oseltamivir,molecular docking,molecular dynamics simulation
Introduction
Neuraminidase has become the main targetfor drug design against influenza virus due to its highly conserved catalytic site and its essential role in influenza virus replication[1-5].Oseltamivir is an antiviraldrug againstneuraminidase thatis usefulforpreventing viral replication in the laststage of the virallive cycle[6].It directly interacts with the catalytic residuesofthe neuraminidase catalytic site,while the framework residues stabilize the enzyme structure[7].Resistance to oseltamivirhas already become a widespread phenomenon,and prediction ofthe neuraminidase structure showsthatthe resistance could be severaltimes higherthan resistance to zanamivir,a neuraminidase inhibitor[8,9].Mutations at the conserved residues of neuraminidase appear to associate with oseltamivirresistancein a subtype specific manner[6].Arg292Lys,Asn294Ser,and His274Tyr are three known types of mutations thatmanifestoseltamivir resistance in H1N1 influenza virus[1,9-11].The frequency of His274Tyrmutantviruses is 64%,although itvaries among differentcountries[9,12].A high prevalence of the mutant virus is seen in South Africa,New Caledonia, New Zealand,the Philippines,and Australia[13],modest in Singapore,Malaysia and Thailand,butinsignificant in Macau and Taiwan[12].
The search for effective drugs that can cope with H1N1 is still ongoing.Li et al.searched zinc fragmentdatabase and found the lead compound of Neo6 as a feasible drug candidate[14].Drug resistance tendecy of oseltamivir towards H5N1 provided valuable data for H1N1 drug design[1,11].Six analog inhibitors as drug candidates against H5N1 were suggested by Du et al.[15].A more robust binding energy than that of zanamivir and oseltamivir was found in compound AG7088 and its derivatives[16,17].
Oseltamivir has good bioavaibility and is effective for treatment and prevention of epidemic influenza infection in adults,adolescents and children(≥one year of age)[18].Modifcation of oseltamivir to cope with influenza Avirus subtype H1N1 has been attempted[1,11], which is based on the properties of amino acid residues in the catalytic site of neuraminidase[19].Drug-target interactions are being investigated using structural bioinformatics tools[20,21].In this study,we focused on the modification of the oseltamivir functionalgroups and molecular docking was performed to determine the bestcandidate ligand based on the lowestbinding energy and interaction.Molecular dynamics simulation is a usefultheoretical tool for analyzing the proteinligand interactions with atomic resolutions based on classicalmechanics[22-25].We also carried outmolecular dynamics simulation to evaluate the interaction ofcandidate ligands and the enzyme in the hydrated state at two differenttemperatures.
Materials and methods
Sequence alignment and homology modeling of neuraminidase
Neuraminidase sequences with mutations were downloaded from National Center for Biotechnology Information(NCBI)influenza virus sequence database (http://www.ncbi.mlm.nih.gov/genomes/flu/).The multiple sequence alignment method was based on the CLUSTAL W2 program(www.ebi.ac.uk/Tools/ clustalw2/index.html)[11,26].The alignment result was forwarded into homology pipeline for further processing of 3D structure determination.The homology modeling was performed using the Swiss model,which can be accessed through http://www.swissmodel.expasy. org/SWISS-MODEL.html.3CKZ chain A[Protein Data Bank(PDB)code]with mutation His274Tyr as template was applied to build the latest structure of N1. Validation ofthe 3Dstructure from the homology modeling was performed by protein geometry program and superimposed by superpose program in MOE 2009.10 software[9].Based on the superimposition,the rootmean-square deviation(RMSD)was calculated to identify structural similarity between template model mutated with 3D structure from homology modeling.
Ligand preparation ofoseltamivir modification
Modification oftheoseltamivirfunctionalgroupswere conducted with alcohol,aldehyde,ketone,carboxylic acid,esther,amide,O-glycoside[27]and apioside group[28]using ACD Labs software[29].Modified oseltamivirwas built into 3D structure using ACD Labs software.3D shape was obtained by saving itin the 3D viewer of ACD Labs.Furthermore,the output format was converted into Molfile MDL Molformatusing software Vegazz to conform the docking process[30].Ligand was washed with a computer program;adjustments were made with the ligand partialcharge and partialcharge optimization using MMFF94x forcefield[9,31].The conformation structure energy of ligands was minimized using the root-mean-square(RMS)gradientenergy with 0.001 kcal/mol[9].Otherparameters were in accordance with existing defaultin the MOE 2009.10 software[32].
Molecular docking
MOE 2009.10 was applied for optimization and minimization of the 3D structure of the enzyme with the addition of hydrogen atoms[9].Protonation was employed with Protonate 3D programs to introduce changes in ionization step.The‘wash’process was started by optimization pipeline,on MOE database viewer.Furthermore,partial charges and force field modification were applied with MMFF94x[9,30].Solvation of enzymes was performed in the form of a gas phase with a fixed charge,RMS gradient of 0.05 kcal/ mol,and other parameters by using the standard in MOE 2009.10 software[9,30,33].
The initialization ofthe docking process was applied with MOE 2009.10[9].Docking simulations were performed by Compute-Simulation dock program.During the docking procedure,ligands were flexible,whereas the receptor was fixed.The application of triangle matcherfor the placementmethod with the iteration of total 1,000,000 energy readings per position and parameter tuning was performed with the standard default value ofMOE[9,33].Atotalof1,000 populationsforscoring function were utilized for LondonDG and repetition force field[9,30,33].The first100 repetitionsand the second setting were shown only as one of the best results.
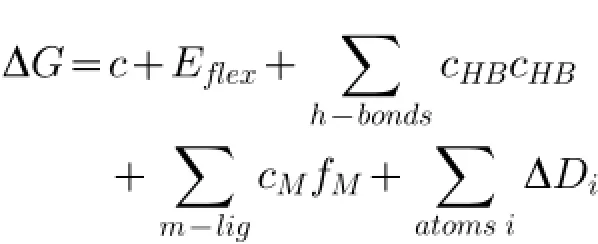
Scoring using London DG estimated Gibbs binding energy(ΔGbinding)from pose of ligand to enzyme was based on the calculation:Where,c=rotation entropy rate and translation thatcan be obtained orreleased,Eflex=flexibility of ligand energy,fHB=imperfection size geometry of hydrogen bonds,cHB=energy of an ideal hydrogen bond,fM=imperfection size geometry of metal ligations,cM=ideal metal ligation energy and Di= atom desolvation energy[31,34-36].The analysis of the ligand interactions with enzyme at both temperatures of 300 K and 312 K was employed by LigX tools in the MOE 2009.10 software.
Molecular dynamics simulation
Molecular dynamics simulation was carried outat2 different temperatures using Simulations-Dynamics program in MOE 2009.10 software[37].The solvation in potentialsetup was born with RMS gradientof0.05[38]. Geometry optimization and energy minimization of 3D structure neuraminidase complex ligandswere perfomed with MOE 2009.10 software[31].Geometry optimization using partial charge was currentforce fields.Energy minimization was employed with MMFF94x.Ensemble parameter was performed:NVT(N,number of atoms; V,volume;T,temperature),NPA algorithm,and Cutoff restraint6A.Dynamics simulation stage involved initialization for 40 ps,main simulations as equilibrium and production stage forproduced trajectory.
Moleculardynamics simulation ofcomplex enzymeligand attemperature of300 Kwasemployed with main simulation for 5,000 ps and cooling for 20 ps[39]. Moleculardynamics simulation attemperature of 312 K was employed from heating attemperature of 300 K to 312 K for 20 ps.Main simulation was employed for 5000 ps and cooling for 20 ps.Position,velocity and acceleration were saved every 0.5 ps.The other parameters were performed in accordance with default MOE dynamics parameters.
Born solvation caused the solvation energy(Esol)to be calculated in potential energy molecular system function from atom coordinate:
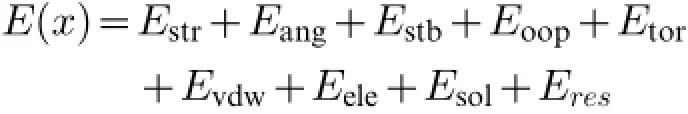
Where,Estr=stretching energy,Eang=angularenergy, Estb=stretching and bending energy,Eoop=operating energy,Etor=energy torsional,Evdw=intermolecules van der Waals energy,Eele=electrostatic energy,Esol=solvation energy,dan Eres=residualmoving energy so thatwe can see clearly Esolin the equation[39-41].
Interaction between ligand and residues after simulation was analyzed using LigX Ligand Interaction in MOE 2009.10 software[42].Visualization of different conformations that occured during simulation was analyzed using Surface and Map program in MOE 2009.10 software.
Results
Homology modeling
Neuraminidase sequence>gi|237651250|gb| ACR08499.1|neuraminidase[Influenza A virus(A/ Auckland/1/2009(H1N1))]was used as the target sequence and had 91.123%similarity with the template of3CKZ chain A(PDB code).Accordingly,the alignmentresults were employed to build homology model for N1 and then superimposed with the template[1]. Homology modeling for neuraminidase of influenza A virus H1N1 was obtained using template 3CKZ chain A that contains mutation His274Tyr and was identified as an oseltamivir-resistantstrain[1,17].
The reliability of homology modelof N1 was identified by Ramachandran plot[11,15].Itwas limited by an orange area,that had coordinate of secondary protein structure as maximum tolerance limit area of steric strain(Fig.1).This area was an allowed region,if there was a protein out of the blue line,putting the amino acid in the outlier(dissallowed region).In the tolerance limit area of residues plot,glysine was not allowed because itdid nothave a side chain,making φ(phi)andψ(psi)angles notlimited.The number of residue plotbesides glysine showed the quality of the protein structure.The results ofthe Ramachandran plotshowed that only Lys331 was in the outlier.About 95.8%of the residues were in the core region of the Ramachandran map,so that the homology model was used for the docking process.Superimposition was used to find out the degree of similarity between template 3CKZ chain A(PDB code)and the homology model. RMSD was calculated to find outthe structural similarity between these proteins.The RMSD value ofsuperimposition was 0.07 A,indicating that the homology model N1 was very close to the structural similarity and was therefore used forthe docking process.
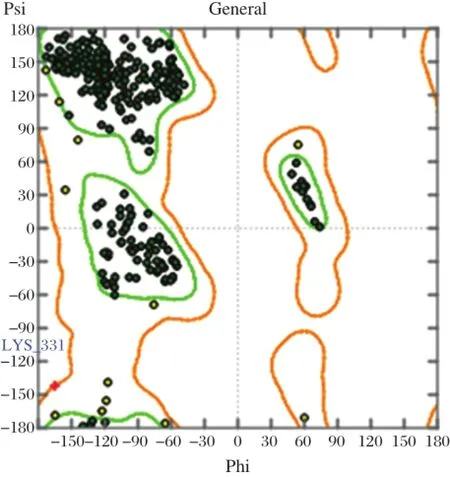
Fig.1 Ramachandran plot of modeling structure of N1.It shows only one outlier Lys331 in dissallowed region.
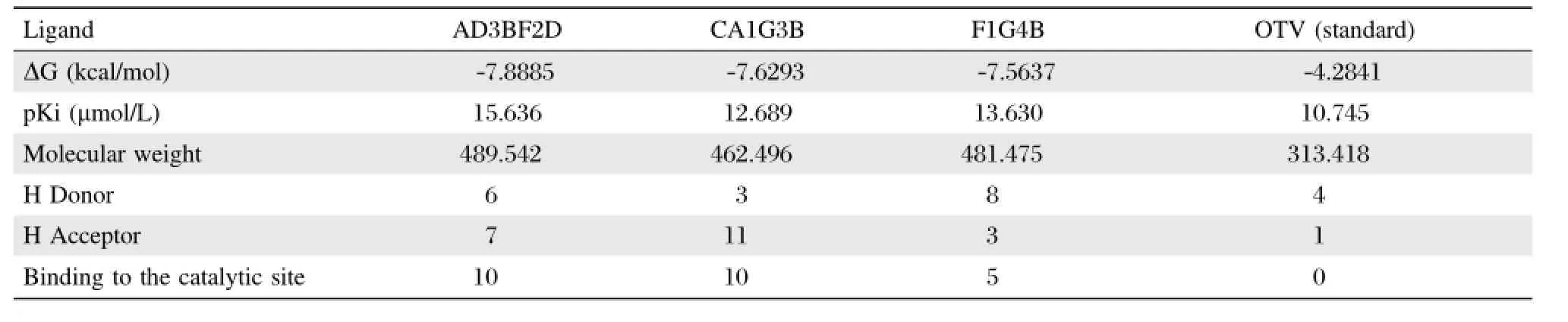
Table 1 The properties of the three best ligands and standard ligand
Screening using molecular docking
A total of 1,232 oseltamivir modified ligands and oseltamivir were screened using molecular docking based on the lowestbinding energy(ΔG)[28].The docking of the 1,232 ligands resulted in 100 best ligands; then,repeated docking resulted in 20 best ligands. Screening was based on Lipinskiˊs rule of five,which showed that the molecular weight for drug likeness must be under 500 g/mol[43,44].
Screening of 20 best ligands resulted in 10 ligands. Those ligands were screened,resulting in three of the bestligands.They were selected based on the quantities of hydrogen to the catalytic site ofneuraminidase. Most of the three best ligands have the O-glycoside and the apioside group as the functionalgroup ofmodified oseltamivir[17,28].These ligands were AD3BF2D, CA1G3B and F1G4B.The apioside group greatly enhanced inhibitory activity28because it is similar to O-glicoside compounds that have inhibitory activity againstneuraminidase,and especially hydrolize influenza virus sialidase[27].
In Table 1,the properties of three ofthe bestligands AD3BF2D,CA1G3B,and F1G4B and oseltamivir as standard ligand showed thattheseligandshad the lowest binding energy than oseltamiviras standards.The level of the lowestΔG value was AD3BF2D,CA1G3B and F1G4B ligands withΔG=-7.8885,-7.6293,and -7.5637 kcal/mol,respectively.These values were better than oseltamivir as the standard ligand withΔG= -4.2841 kcal/mol.pKivalues of these ligands were 15.636,12.689,and 13.630μM,respectively.These values were better than pKivalues of oseltamivir with 10.745μM.These pKi values of AD3BF2D,CA1G3B, and F1G4B ligands indicated thatthese ligands were effective and had betteraffinity to strongly interactwith neuraminidase than oseltamivir.The molecular weights of these ligands were under 500 g/mol.AD3BF2D, CA1G3B and F1G4B ligands had a numberofbindings with the catalytic site(10,10 and 5 bindings,respectively)(Table 1).These ligand bindings in the catalytic site were chosen for molecular dynamics simulation as oseltamvir had fewer bindings with the catalytic site[28,45].
Hydrogen bondsbetween amino acid residuesofneuraminidase with AD3BF2D,CA1G3Band F1G4B ligands and oseltamivir are shown in Table 2.AD3BF2D, CA1G3B and F1G4B ligands had more binding affinity with the catalytic site than oseltamivir.AD3BF2D ligand had hydrogen bonds with Arg118,two bindings with Glu278 and Arg 293,two bindings with Arg368 and fourbindingswith Tyr402.CA1G3B ligand had hydrogen bonds with Arg118 and Asp151,three bindings with Arg293,two bindingswith Arg368 and three bindings with Tyr402.F1G4B ligand has hydrogen bondswith Asp151,two bindings with Glu278 and two bindings with Tyr402.However,oseltamivir did nothave hydrogen bond forinteracting with the catalytic site of neuraminidase.The quantities of the interacting hydrogen bondswith the catalytic site ofneuraminidase indicated that these ligands have the ability to inhibitneuraminidase.Ligands AD3BF2D,CA1G3B and F1G4B had many hydrogen bond interactions with the catalytic site of neuraminidase.The docking poses in N1 of these three ligands are shown in Fig.1.These hydrogen bonds were chosen as the basis of molecular dynamics simulation.

Table 2 Interactions of three of the best ligands and standard ligands with neuraminidase after docking
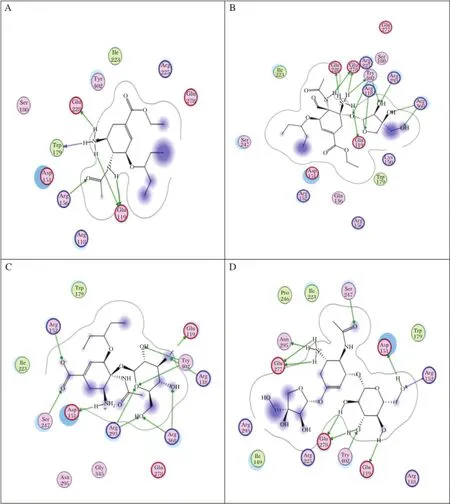
Fig.2 The docking processes of the three best ligands in N1.A:OTV,B:AD3BF2D,C:CA1G3B,and D:F1G4B.
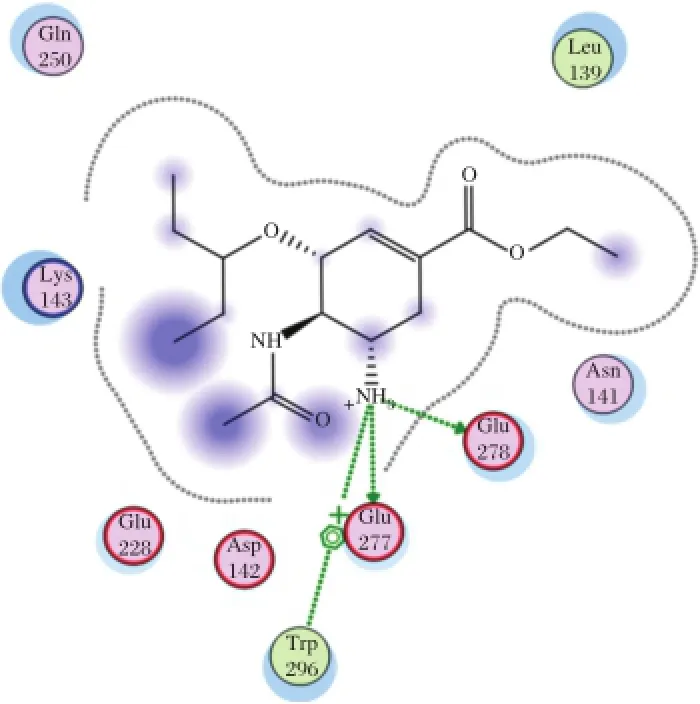
Fig.3 Interaction to form hydrogen bonds of OTV ligand at the end simulations of temperature of 300 K.
Molecular dynamics simulation
The evolution time of the hydrogen bonds from the inhibitor-enzyme complex provides an approach to evaluate the convergence of the dynamicalproperties of the system[25].The interaction of the bestligands, AD3BF2D,CA1G3B and F1G4B with the enzyme was evaluated in the hydrated state using molecular dynamics simulation at two different temperatures (300 K and 312 K).The simulation at temperature of 300 K was chosen to evaluate the interaction of the complex enzyme-ligand atthe room temperature.
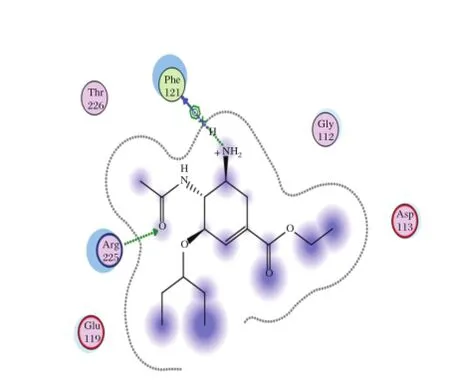
Fig.4 Interaction to form hydrogen bonds of OTV ligand at the end simulations of temperature of 312 K.
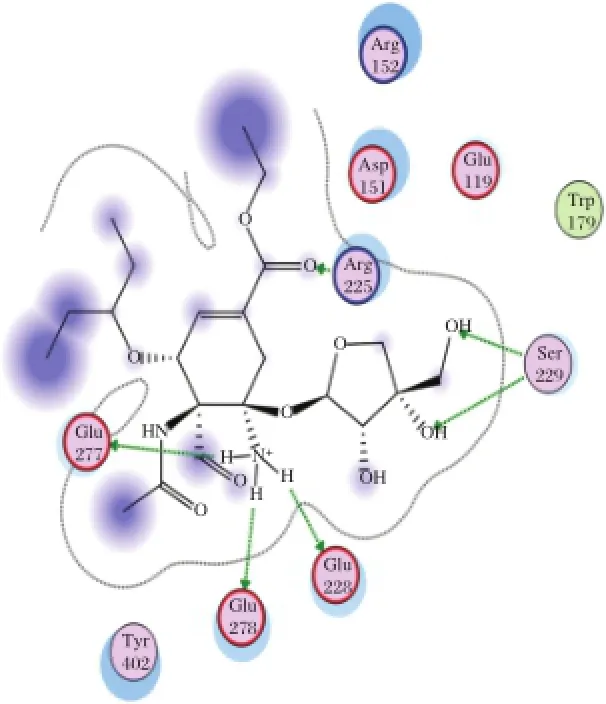
Fig.5 Interaction to form hydrogen bonds of AD3BF2D ligand at the end simulations of temperature of 300 K.
The simulation attemperature of312 Kwaschosen to evaluate the interaction of the complex enzyme-ligand during the fever.As shown in Table 3,AD3BF2D has good interaction than the other ligands,CA1G3B and F1G4B.Compared with oseltamiviras the standard ligand(Fig.2and 3),AD3BF2D had better interaction in two simulations attemperature of300 K and 312 K. The interaction of AD3BF2D to form hydrogen bonds was much more stable in molecular docking,until the end ofthe simulations.However,the interaction between molecular docking and molecular dynamics simulation decreased from 10 hydrogen bonds to 1 and 3 hydrogen bondsatthe end ofsimulationsoftemperaturesof300 K and 312 K,respectively.
The stability of hydrogen bonds occured between Glu278 as one ofthe catalytic residues ofneuraminidase and NH3as the functional group of AD3BF2D ligand during simulations at temperature of 300 K(Fig.4).
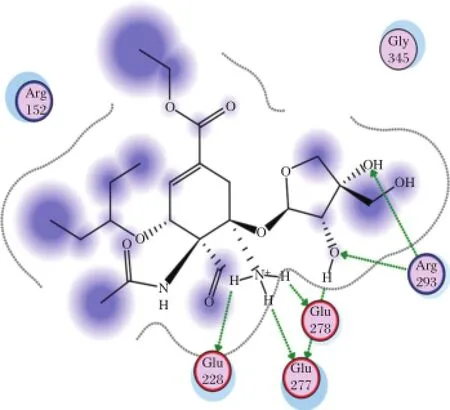
Fig.6 Interaction to form hydrogen bonds of AD3BF2D ligand at the end simulations of temperature of 312 K.
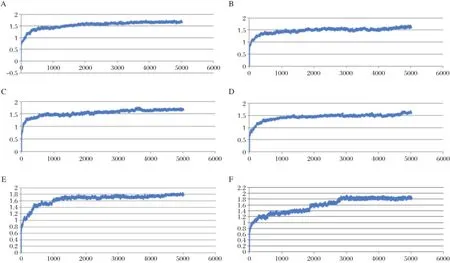
Fig.7 The RMSD Graphic of(A)CA1GB and Neuraminidase(NA)dynamics at 300 K.The X axis represents the molecular dynamics time duration in picoseconds(ps),while the Y axis represents the RMSD value.The graphics shows that after 2,000 ps,the structure become stable. B:The RMSD Graphic of CA1GB and Neuraminidase(NA)dynamics at312 K.The graphics shows thatafter2,000 ps,the structure become stable.C: The RMSD Graphic of F1G4B and Neuraminidase(NA)dynamics at300 K.The graphics shows thatafter 2,000 ps,the structure become stable.D: The RMSD Graphic of F1G4B and Neuraminidase(NA)dynamics at 312 K.The graphics shows that after 2,000 ps,the structure become stable. E:The RMSD Graphic of AD3BF and Neuraminidase(NA)dynamics at300 K.The graphics shows thatafter3,000 ps,the structure become stable.F: The RMSD Graphic of AD3BF2D and Neuraminidase(NA)dynamics at312 K.The graphics shows thatafter 3,000 ps,the structure become stable.
The stability of hydrogen bonds also occured between Glu278 and NH3 functionalgroup of AD3BF2D during simulations at 312 K.The stability of hydrogen bonds was also increased at simulations at 312 K between Arg293 and alcoholofthe apioside group asthefunctional group of AD3BF2D ligand;and this interaction with Arg293 as catalytic residue ofneuraminidase resulted in two hydrogen bonds(Fig.5).Ifcompared with the interaction thathas 10 hydrogen bonds after docking,in the binding catalytic site,interaction after simulations could notform hydrogen bonds due to the mobility of amino acid residues in the catalytic site.Ithad differentmotions in solvent or hydrated conditions.These motions proved thatthere were dynamic enzymesdue to the effectofsolventmolecules.The presence of the differentfunctional groups in each ligand had various bond effects that resulted in the differentconformations.The stable interaction of AD3BF2D ligand to form hydrogen bonds during simulations attwo differenttemperatures provided solid evidence thatthis ligand had affinity with neuraminidase. More complete ligand interactionsdata are in the supplementary material(http://www.bioinf.uni-leipzig.de/~arli/supplementary-material.pdf).As shown in Fig.6,there were changes in conformation of the enzyme-AD3BF2D ligand complex,which occured during simulation,and showed the dynamic behaviour of the enzyme in the presence of solventand ligand. AD3BF2D ligand remained in the binding pocketafter docking,initialization stage and atthe end of simulations at 300 K and 312 K.The binding pocket of this complex changed from the‘closed’form into the‘open’state atthe end ofsimulation.Based on the simulation results,the changes in conformation of enzyme-AD3BF2D ligand complex during simulations caused apioside and ammonium functionalgroups to form more stable interactions with Glu278 and Arg293 compared with others.Thosefunctionalgroups,namely pentyloxy, aldehyde,and carboxylate did notform hydrogen bonds with binding pocket.More conformation figuresofother ligands are listed in the Supplementary Material). Fig.7A-F shows thatthe ligand-receptor interaction indeed reached its stability after 2,000-3,000 ps.This would suggestthatthe ligands,as drug candidates,are within the stable condition in the human body.
Discussion
In this research,the contribution from functional groups AD3BF2D ligand(N-[(1S,6R)-5-amino-5-{[(2R,3S,4S)-3,4-dihydroxy-4-(hydroxymethyl)tetrahydrofuran-2-yl]oxy}-4-formylcyclohex-3-en-1-yl] acetamide-3-(1-ethylpropoxy)-1-cyclohexene-1-carboxylate),apioside and ammonium group provide better interaction to form hydrogen bonds as affinity to neuraminidase than oseltamivir due to the polarity of functional groups,so that these functional groups are able to form interaction with neuraminidase thathave polar and hydrophilic residues in the catalytic site. The apioside group forms two hydrogen bonds with Arg293,and the ammonium group forms one hydrogen bond with Glu278 as a residue in the catalytic site of neuraminidase.This resultofinteraction ofthe apioside and ammonium group with the residue in the catalytic site of neuraminidase indicated that AD3BF2D ligand can inhibitneuraminidase and this ligand can be proposed as a candidate for neuraminidase inhibitor of influenza A virus subtype H1N1.
As an outlook of this study,we are aware thatsome research groups already produced RNA based drugs for interfering with influenza virus[46-48].Moreover,a feasible and powerful transcriptomics based tool has been generated for viral bioinformatics research[49-52].To this end,itwould be interesting to investigate the transcriptomics properties ofthe H1N1 virus,as supplementary approach to the existing proteomics method.
In conclusion,we have built the latest N1 structure model by homology modeling,which has excellent reliability by using Ramachandran plot and template superimposition.A totalof1,232 oseltamivir modified ligands molecules have been designed and screened by moleculardocking.Three ofthe bestligands,AD3BF2D, CA1G3B,and F1G4B were obtained with the lowest binding energy thatcontradicted to OTV as standard. These ligandsˊinteractions were evaluated in hydrated state using molecular dynamics simulation.This work has provided AD3BF2D ligands thathave stable interaction with Glu278 and Glu 278,Arg293 and Arg 293 during simulations at temperature of 300 K and 312 K, respectively.Based on these results,AD3BF2D ligand can be used as a candidate for neuraminidase inhibitor againstinfluenza virus A subtype H1N1.
Acknowledgements
The authors would like to thank Hibah BOPTN PUPT Ditjen Dikti No:0970/H2.R12/HKP.05.00/2014 and Directorate of Research and Community Engagement,University of Indonesia,for supporting this research.Usman Sumo Friend Tambunan supervised this research,Rizky Archintya Rachmania was working on the technicaldetails and preparing the research report and Arli Aditya Parikesit was responsible for writing and revising the manuscript.
[1]Wang S-Q,Du Q-S,Huang R-B,et al.Insights from investigating the interaction of oseltamivir(Tamiflu)with neuraminidase of the 2 00 9 H1 N1 swine flu v irus. Biochem Biophys Res Commun 2009;386(3):432-436.
[2]Varghese JN,Epa VC,Colman PM.Three-dimensional structure of the complex of 4-guanidino-Neu5Ac2en and influenza virus neuraminidase.Protein Sci 1995;4(6): 1081-1087.
[3]Wang MZ,Tai CY,Mendel DB.Mechanism by which mutations at his274 alter sensitivity of influenza a virus n1 neuraminidase to oseltamivir carboxylate and zanamivir.Antimicrob Agents Chemother.2002;46(12):3809-3816.
[4]Liu A,Cao H,Du G.Drug screening for influenza neuraminidase inhibitors.Sci China C Life Sci 2005;48(1):1-5.
[5]Wen W-H,Wang S-Y,TsaiK-C,etal.Analogs ofzanamivir with modified C4-substituents as the inhibitors against the group-1 neuraminidases of influenza viruses.Bioorg Med Chem 2010;18(11):4074-4084.
[6]Rungrotmongkol T,Intharathep P,Malaisree M,et al. Susceptibility of antiviral drugs against 2009 influenza A(H1N1)virus.Biochem Biophys Res Commun 2009; 385(3):390-394.
[7]Ferraris O,Lina B.Mutations of neuraminidase implicated in neuraminidase inhibitors resistance.J Clin Virol 2008;41(1):13-19.
[8]Moscona A.Global transmission of oseltamivir-resistant influenza.N Engl J Med 2009;360(10):953-956.
[9]Rosmalena,Fadhilah,Tedjo A.Design of Linear Peptide as Neuraminidase Inhibitor Influenza A Virus Base on Molecular Docking Simulation.Proc Third Int Conf Math Nat Sci(ICMNS 2010)2010.
[10]Chen C-YCY-C,Huang H-J,Tsai F-J.Drug design for Influenza A virus subtype H1N1.J Taiwan Inst Chem Eng 2010;41(1):8-15.
[11]Wei D-Q,Du Q-S,Sun H,et al.Insights from modeling the 3D structure of H5N1 influenza virus neuraminidase and its binding interaction s with ligands.Biochem Biophys Res Commun 2006;344(3):1048-1055.
[12]Hurt AC,Ernest J,Deng Y-M,et al.Emergence and spread of oseltamivir-resistant A(H1N1)influenza viruses in Oceania,South East Asia and South Africa.Antiviral Res 2009;83(1):90-93.
[13]Besselaar TG,Naidoo D,Buys A,etal.Widespread oseltamivir resistance in influenza A viruses(H1N1),South Africa.Emerg Infect Dis 2008;14(11):1809-1810.
[14]Li X-B,Wang S-Q,Xu W-R,etal.Novelinhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method.PLoS One 2011;6(11):e28111.
[15]Du Q-S,Wang S-Q,Chou K-C.Analogue inhibitors by modifying oseltamivir based on the crystal neuraminidase structure for treating drug-resistant H5N1 virus.Biochem Biophys Res Commun 2007;362(2):525-531.
[16]Patick AK,Binford SL,Brothers MA,et al.In Vitro Antiviral Activity of AG7088,a Potent Inhibitor ofHuman Rhinovirus 3C Protease.Antimicrob Agents Chemother 1999;43(10):2444-2450.
[17]Tambunan USF,AmriN,Parikesit AA.In silico design of cyclic peptides as influenza virus,a subtype H1N1 neuraminidase inhibitor.African J Biotechnol 2012;11(52): 11474-11491.
[18]Ward P,Small I,Smith J,etal.Oseltamivir(Tamiflu)and its potentialforuse in the eventof an influenza pandemic. J Antimicrob Chemother 2005;55 Suppl 1:i5-i21.
[19]Yen H-L,Hoffmann E,Taylor G,et al.Importance of neuraminidase active-site residues to the neuraminidase in h ib ito r resistan ce o f influ enza v iru ses.J Virol 2006;80(17):8787-8795.
[20]Chou K-C.Structural bioinformatics and its impact to biomedical science.Curr Med Chem 2 00 4;1 1(16): 2105-2134.
[21]Chou K-C,Wei D-Q,Zhong W-Z.Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS.Biochem Biophys Res Commun 2003;308(1):148-151.
[22]Adcock SA,McCammon JA.Molecular dynamics:survey of methods for simulating the activity of proteins.Chem Rev 2006;106(5):1589-1615.
[23]Van Gunsteren WF,Bakowies D,Baron R,et al. Biomolecular modeling:Goals,problems,perspectives. Angew Chem Int Ed Engl 2006;45(25):4064-4092.
[24]Ravna AW,Sylte I,Dahl SG.Structure and localisation of drug binding sites on neurotransmitter transporters.J Mol Model 2009;15(10):1155-1164.
[25]Wang S-Q,Cheng X-C,Dong W-L,et al.Three new powerful oseltamivir derivatives for inhibiting the neuraminid ase of in fluenza v iru s.Biochem Biophys Res Commun 2010;401(2):188-191.
[26]Wang P,Hu L,Liu G,et al.Prediction of antimicrobial peptides based on sequence alignment and feature selection methods.PLoS One 2011;6(4):e18476.
[27]Guo C-T,Sun X-L,Kanie O,et al.An O-glycoside of sialic acid derivative that inhibits both hemagglutinin and sialidase activities of influenza viruses.Glycobiology 2002;12(3):183-190.
[28]Ryu YB,Kim JH,Park S-J,et al.Inhibition of neuraminidase activity by polyphenol compounds isolated from the roots of Glycyrrhiza uralensis.Bioorg Med Chem Lett 2010;20(3):971-974.
[29]Spessard GO.ACD Labs/LogP dB 3.5 and ChemSketch 3.5. J Chem Inf Model1998;38(6):1250-1253.
[30]Sunaryo H,Rachmania RA.Screening of Bioactive Compounds from Olea Europaea as Glutamine Synthase A Inhibitor of Bacterial Meningitis haemophilus Infulenza Type B Through Molecular Docking Simulation.Proceeding Int Conf 2nd Pharm Adv Pharm Sci 2011.
[31]Tambunan USF,Harganingtyas R,Parikesit AA.In silico Modification of(1R,2R,3R,5S)-(-)-Isopinocampheylamine as Inhibitors of M2 Proton Channel in Influenza A Virus Subtype H1N1,using the Molecular Docking Approach. Trends Bioinforma 2012;5(2):25-46.
[32]Vilar S,Cozza G,Moro S.Medicinal chemistry and the molecular operating environment(MOE):application of QSAR and molecular docking to drug discovery.Curr Top Med Chem 2008;8(18):1555-1572.
[33]Tambunan USF,Fadilah F,Parikesit AA.Bioactive Compounds Screening from Zingiberaceae Family as Influenza A/Swine Flu Virus Neuraminidase Inhibitor through Docking Approach.Online J Biol Sci 2010; 10(4):151-156.
[34]Naı¨m M,Bhat S,Rankin KN,et al.Solvated interaction energy(SIE)for scoring protein-ligand binding affinities. 1.Exploring the parameter space.J Chem Inf Model 47(1):122-133.
[35]Wojciechowski M,Lesyng B.Generalized Born Model: Analysis,Refinement,and Applications to Proteins.J Phys Chem B 2004;108(47):18368-18376.
[36]Galli CL,Sensi C,Fumagalli A,et al.A computational approach to evaluate the androgenic affinity of iprodione, procymidone,vinclozolin and their metabolites.PLoS One 2014;9(8):e104822.
[37]Johnson BC,Metifiot M,Pommier Y,et al.Molecular dynamics approaches estimate the binding energy of HIV-1 integrase inhibitors and correlate with in vitro activity.Antimicrob Agents Chemother 2012;56(1):411-419.
[38]Tamb un an USF,Apriyanti N,Parikesit AA,et al. Computational design of disulfide cyclic peptide as potential inhibitor of complex NS2B-NS3 dengue virus protease.African J Biotechnol 2011;10(57):12281-12290.
[39]Tambunan USF,Noors RS,Parikesit AA.Molecular Dynamics Simulation of DENV RNA-Dependent RNAPolymerase with Potential Inhibitor of Disulfide Cyclic Peptide.Online J Biol Sci 2011;11(2):48-62.
[40]Neria E,Nitzan A.Simulations of solvation dynamics in simple polar solvents.J Chem Phys 1992;96(7):5433.
[41]Tambunan USF,Parikesit AA.HPV Bioinformatics:In Silico Detection,Drug Design and Prevention Agent Development.In:Rajkumar R,ed.Topics on Cervical Cancer with an Advocacy for Prevention Rijeka,Croatia, Croatia:Intech Publishing;2012:237-252.
[42]Sarker S,Weissensteiner R,Steiner I,etal.The high-affinity binding site for tricyclic antidepressants resides in the outervestibule of the serotonin transporter.MolPharmacol 2010;78(6):1026-1035.
[43]Tambunan USF,Parikesit AA,Hendra,et al.In Silico Analysis of Envelope Dengue Virus-2 and Envelope Dengue Virus-3 Protein as the Backbone of Dengue Virus Tetravalent Vaccine by Using Homology Modeling Method.Online J Biol Sci2009;9(1):6-16.
[44]Lipinski CA,Lombardo F,Dominy BW,et al.Experimentaland computationalapproaches to estimate solubility and permeability in drug discovery and development settings.Adv Drug Deliv Rev 1997;23(1-3):3-25.
[45]Tambunan USF,Bramantya N,Parikesit AA.In silico modification of suberoylanilide hydroxamic acid(SAHA) as potential inhibitor for class II histone deacetylase (HDAC).In:Ranganathan S,ed.BMC bioinformatics Vol 12 Suppl1.BioMed Central Ltd;2011:S23.
[46]Bonetta L.RNA-based therapeutics:ready fordelivery?Cell 2009;136(4):581-584.
[47]Castanotto D,RossiJJ.The promises and pitfalls of RNA-interference-based therapeutics.Nature 2009;457(7228): 426-433.
[48]Stevenson M.Therapeutic Potential of RNA Interference. N Engl J Med 2004;351:1772-1777.
[49]HofackerIL,Fekete M,Flamm C,etal.Automatic detection of conserved RNA structure elements in complete RNA virus genomes.Nucleic Acids Res 1998;26(16):3825-3836.
[50]Thurner C,Witwer C,Hofacker IL,Stadler PF.Conserved RNA secondary structures in Flaviviridae genomes.J Gen Virol 2004;85(Pt 5):1113-1124.
[51]Hofacker IL,Stadler PF.Automatic detection of conserved base pairing patterns in RNA virus genomes. Comput Chem 1999;23(3-4):401-414.
[52]Hofacker IL,Fekete M,Stadler PF.Secondary structure prediction for aligned RNA sequences.J Mol Biol 2002; 319(5):1059-1066.
✉Correspo nding author:Usman Sumo Friend Tambu nan, Bioinformatics Research Group,Department of Chemistry,Faculty of Mathematicsand NaturalScience,University ofIndonesia,Depok Campus, Depok 16424,Indonesia.Tel/Fax:+6221 7270027/+6221 7863432,
E-mail:usman@ui.ac.id.
Received 01 May 2013,Revised 26 June 2013,Accepted 18 August2014, Epub 12 December 2014
The authors reported no conflict of interests.
©2015 by the Journal of Biomedical Research.All rights reserved.
10.7555/JBR.29.20130024
 THE JOURNAL OF BIOMEDICAL RESEARCH2015年2期
THE JOURNAL OF BIOMEDICAL RESEARCH2015年2期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Recent advances in bariatric/metabolic surgery:appraisalof clinical evidence
- Metabolic bariatric surgery and type 2 diabetes mellitus:an endocrinologistˊs perspective
- Roux-en-Y gastric bypass for Chinese type 2 diabetes mellitus patients with a BMI<28 kg/m2:a multi-institutionalstudy
- Bariatric surgery in old age:a comparative study of laparoscopic Roux-en-Y gastric bypass and sleeve gastrectomy in an Asia centre ofexcellence
- Maternalinheritance of severe hypertriglyceridemia impairs glucose metabolism in offspring
- Osthole inhibits proliferation of human breast cancer cells by inducing cell cycle arrest and apoptosis