
痕量单一价态钚溶液的制备
2014-12-25 07:47:26韩小元张瑞荣吴王锁
核化学与放射化学 2014年4期
王 煜,韩小元,张瑞荣,周 旭,刘 艳,吴王锁
1.兰州大学 放射化学研究所,甘肃 兰州 730000;2.西北核技术研究所,陕西 西安 710024
钚是一种极毒的放射性元素,高放废物处置库建设需研究钚的环境化学行为。钚的环境化学行为受配位、沉淀、吸附、氧化还原、胶体存在等条件的影响[1-2]。其中,氧化还原作用对钚的溶解和迁移行为影响最大[3]。钚在水文地质环境中可同时以4种价态存在(Pu(Ⅲ)、Pu(Ⅳ)、Pu(Ⅴ)、Pu(Ⅵ)),其中,在氧化性地表水体系中以四、五、六价为主[4],四价钚的吸附能力极强且溶解度极低,易吸附到固相表面或以胶体态存在,而溶解态的钚一般以五价存在[5-6],同时,钚与环境介质作用过程中不同价态之间还可以相互转化[7-8]。此外,对钚在污染场地地下水中的迁移研究表明,钚浓度一般为超痕量到痕量水平(例如,美国内华达试验场中,距地下核试验爆心1.3km处地下水239Pu浓度为10-14mol/L[2])。因此,对不同价态痕量钚的环境化学行为研究更接近于实际环境[9-11]。
对痕量单一价态钚的环境化学行为研究中,首先必须制备无支持氧化还原剂的痕量单一价态钚溶液。常用单一价态钚溶液的制备方法有电解法[12]和氧化还原法[7-9,13-14]。电解法通过控制电解电位,可获得高纯度单一价态钚溶液,且不会引入其他杂质,但只适用于高浓度钚溶液的制备。氧化还原法通过选择合适氧化还原剂制备单一价态钚溶液[14]。Pu(Ⅳ)的制备较简单,一般在HNO3溶液中反复蒸干即可获得[14]。Pu(Ⅵ)一般用HClO4、KMnO4、NaBrO3氧化的方法制得[7-9,13],然后用化学方法去除氧化剂。Pu(Ⅴ)可利用Pu(Ⅵ)经自还原反应制得[7-8]。对痕量单一价态钚溶液的直接制备未见报道,本研究建立了痕量单一价态钚溶液的制备方法,同时研究了各价态随时间变化的稳定性。
1 实验部分
1.1 仪器与试剂
1220QUANTULUSTM型液体闪烁谱仪,美国PE公司;S20P型pH计,瑞士METTLER TOLEDO公司;H2Q-C空气浴振荡器,常州诺基仪器公司。238Pu、239Pu、242Pu储备液,西北核技术研究所提供;噻吩甲酰三氟丙酮(TTA)、二(2-乙基己基)磷酸(HDEHP)、1-苯基-3-甲基-4-苯甲酰基吡唑啉酮-5(PMBP),均为分析纯,美国Sigma-Aldrich公司;二甲苯,市售分析纯;其它试剂均为市售优级纯。
1.2 实验方法
1.2.1 钚价态分析方法
(1)TTA萃取Pu(Ⅳ)
在5mL聚乙烯塑料管中,加入0.7mL待分析溶液,用0.3mL 3.3mol/L HNO3调节为1mol/L HNO3体系,加1mL 0.5mol/L TTA-二甲苯,300r/min振荡萃取15min,1 000r/min离心2min,取水相0.7mL加入3mL Safe3闪烁液,液闪谱仪测量水相中钚的含量(Pu(Ⅴ)+Pu(Ⅵ))。
(2)PMBP萃取Pu(Ⅳ)
将待分析溶液调节为1mol/L HNO3体系,1mL 0.1mol/L PMBP-二甲苯萃取后,离心分离,液闪谱仪测量水相中钚的含量(Pu(Ⅴ)+Pu(Ⅵ))。
(3)HDEHP萃取Pu(Ⅳ)+Pu(Ⅵ)
将待测液0.7mL用0.3mL 3.3mol/L HCl调节为1mol/L HCl体系,加1mL 0.5mol/L TTA-二甲苯萃取,离心分离,液闪谱仪测量水相中钚的含量(Pu(Ⅴ))。
(4)LaF3沉淀
取待测液1mL,调节体系为1mol/L HNO3、0.5mol/L H2SO4、0.02mol/L Na2Cr2O7,加2g/L La3+、1g/L Zr4+作为共沉淀剂,调节HF浓度为2mol/L,总体积为2mL,震荡2min,离心;用2mL(其 中 含1mol/L HNO3、0.5mol/L H2SO4、0.02mol/L Na2Cr2O7、2mol/L HF)分2次洗涤沉淀,沉淀用1mL 1mol/L HNO3/饱和H3BO3溶解,液闪谱仪测量沉淀溶解物中钚(Pu(Ⅲ)+Pu(Ⅳ))含量,在氧化性环境条件下,Pu(Ⅲ)的存在可基本忽略,因此,实验沉淀部分可视为Pu(Ⅳ)。
1.2.2 Pu(Ⅳ)溶液的制备及稳定性 取2.5×10-12mol/L的238Pu储 备 液(8mol/L HNO3体系)50mL过Dowex1×4阴离子交换柱,依次用10mL 8mol/L HNO3、2mL 1mol/L HNO3淋洗除杂,30mL 0.01mol/L HNO3-0.001mol/L HF解吸钚,将解吸液蒸至近干,加1mL 1mol/L HNO3反复蒸干3次,用10mL 0.01mol/L NaCl溶液溶解,调节pH=3,将制备的Pu(Ⅳ)溶液避光保存40d,测试钚浓度及价态变化。
1.2.3 Pu(Ⅵ)和Pu(Ⅴ)溶液的制备 将钚解吸液蒸至近干,调节溶液为0.8mol/L HNO3体系,KMnO4浓度为0.000 1mol/L。铝箔包裹避光保存24h,此时,钚被氧化为Pu(Ⅵ)。用10mL 0.01mol/L NaCl稀释,调节pH≈2.5,加入与KMnO4等量的MnCl2沉淀,经0.45μm微孔滤膜过滤到聚四氟乙烯瓶中,用NaOH调节pH=3.0,避光保存5d,Pu(Ⅵ)被还原为Pu(Ⅴ)。
2 结果与讨论
2.1 钚价态分析方法选择
2.1.1 萃取剂选择及最佳萃取时间的确定 TTA、PMBP可选择性萃取Pu(Ⅳ),HDEHP萃取Pu(Ⅳ)和Pu(Ⅵ),从而得出溶液中的Pu(Ⅳ)、Pu(Ⅴ)、Pu(Ⅵ)的分布,该萃取方法已与光谱学方法进行比对,偏差小于5%[15]。不同萃取剂所需萃取时间不同,同时,为避免萃取时钚价态可能发生的变化,应尽量缩短萃取时间,因此,需由实验确定最佳萃取平衡时间。钚的萃取比例随时间变化示于图1。由图1可见,使用TTA和HDEHP萃取时,需15min达到萃取平衡,而PMBP在5min即可达平衡,但PMBP溶解性能较差。因此,实验选择TTA为Pu(Ⅳ)的萃取剂,HDEHP为Pu(Ⅳ)+Pu(Ⅵ)的萃取剂,萃取时间均选择15min。
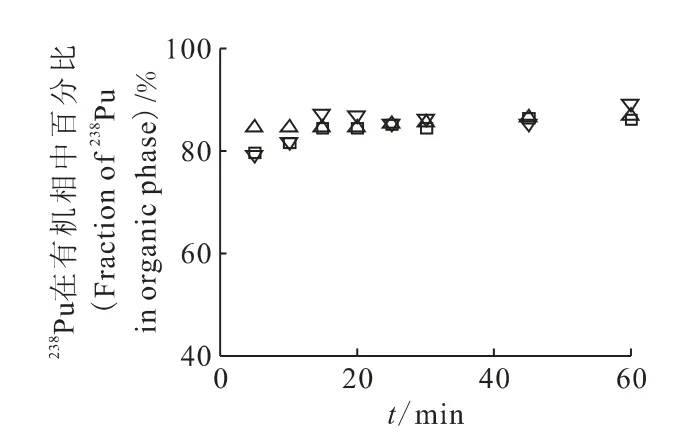
图1 钚在有机相中的百分比随萃取时间的变化Fig.1 Fraction of plutonium in organic phase at different extraction equilibrium time
2.1.2 萃取法与沉淀法结果比对 分别采用TTA-HDEHP萃取法和LaF3沉淀法分析所制备的Pu(Ⅳ)溶液中各价态钚的浓度。TTAHDEHP萃取法分析得出各价态钚的份额为:Pu(Ⅳ)为(96.3±1.5)%、Pu(Ⅴ)为(3.2±0.5)%、Pu(Ⅵ)为(0.5±1.8)%(n=3);LaF3沉淀法分析结果为:Pu(Ⅳ)为(93.2±2.3)%(n=3)。采用F检验和t检验法对两种方法的分析结果进行检验,结果表明,在95%置信度下,两种方法之间无显著性差异,均可用于钚价态分布的测定。
2.2 Pu(Ⅳ)溶液的制备及稳定性
使用1.2节方法制备Pu(Ⅳ)溶液,所制溶液中238Pu含量为1.2×10-11mol/L,Pu(Ⅳ)份额为(96.3±1.5)%。将制备的溶液在pH=3.0条件下保存40d,监测钚浓度及价态分布变化,结果列于表1。由表1可见,在pH=3.0体系中,随着时间延长,钚总量逐渐减少,四价钚易被容器吸附或形成钚胶体,而五、六价钚基本不会被吸附[11]。因此,为了避免钚的损失,实验所用的Pu(Ⅳ)溶液应新鲜制备。
2.3 Pu(Ⅵ)溶液的制备及稳定性
在Pu(Ⅵ)溶液的制备中,需将钚从Pu(Ⅳ)氧化到Pu(Ⅵ),本工作采用KMnO4氧化法制备Pu(Ⅵ)溶液,研究了KMnO4浓度及酸体系、酸浓度、反应时间对氧化效果的影响。

表1 pH=3.0,c(NaCl)=0.01mol/L溶液中238Pu(Ⅳ)的稳定性Table 1 Stabilization of 238Pu(Ⅳ)at pH=3.0,c(NaCl)=0.01mol/L
2.3.1 KMnO4浓度及酸体系的影响 为了将钚由Pu(Ⅳ)氧化到Pu(Ⅵ),需选择合适的KMnO4浓度及酸体系。将1.05×10-11mol/L238Pu(Ⅳ)溶液调节到不同酸体系和KMnO4浓度,避光反应18h,TTA萃取法分析钚价态,计算Pu(Ⅳ)被氧化为Pu(Ⅴ+Ⅵ)的份额。结果示于图2。由图2可见,在硝酸体系中,当KMnO4浓度低至0.1mmol/L时,Pu(Ⅳ)仍能被氧化,增大KMnO4浓度对氧化效果影响不大,且可能在后续去除时造成钚的损失;HCl溶液体系中,KMnO4会被HCl还原,影响其对钚的氧化效果。因此,Pu(Ⅳ)氧化到Pu(Ⅵ)的溶液体系选为0.1mmol/L KMnO4-1mol/L HNO3。
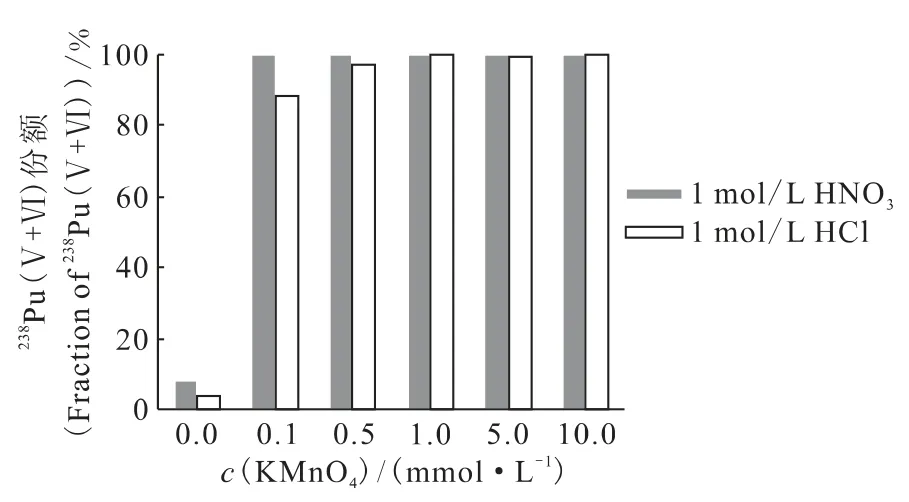
图2 不同KMnO4浓度及酸体系中Pu(Ⅴ+Ⅵ)的份额Fig.2 Fraction of Pu(Ⅴ+Ⅵ)at different concentration of KMnO4and acid system
2.3.2 酸度的影响 在Pu(Ⅳ)溶液中加入不同浓度HNO3,避光反应18h,钚被氧化的份额示于图3。由图3可见,当HNO3浓度为0.5~1.0mol/L时,Pu(Ⅳ)均可被完全氧化;当HNO3浓度大于1mol/L时,Pu(Ⅴ+Ⅵ)份额逐渐减少。Pu(Ⅳ)被氧化为Pu(Ⅴ)反应式为:5Pu(Ⅳ)++6H2O→5Pu(Ⅴ+Mn2++12H+,Pu(Ⅵ)的生成反应与此式类似,由式可见,c(H+)增大时,反应平衡左移,不利于Pu(Ⅴ+Ⅵ)的生成。同时,高浓度HNO3能保持钚在四价,亦不利于钚的氧化。因此,Pu(Ⅳ)氧化到Pu(Ⅵ)时需控制溶液HNO3浓度为0.5~1.0mol/L。

图3 不同硝酸浓度时溶液中Pu(Ⅴ+Ⅵ)的份额Fig.3 Fraction of Pu(Ⅴ+Ⅵ)at different concentration of HNO3
2.3.3 反应时间的影响 溶液中Pu(Ⅴ+Ⅵ)的份额随反应时间变化关系示于图4。由图4可见,随着反应时间延长,溶液中Pu(Ⅴ+Ⅵ)逐渐增多,当反应时间大于16h时,溶液中Pu(Ⅴ+Ⅵ)总量趋于稳定。为了溶液中Pu(Ⅳ)被完全氧化,选择反应时间为24h。
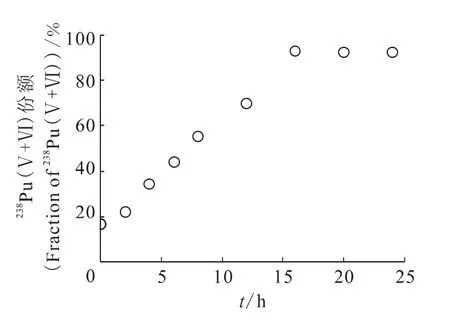
图4 不同反应时间时溶液中Pu(Ⅴ+Ⅵ)的份额Fig.4 Fraction of Pu(Ⅴ+Ⅵ)at different reaction time
2.3.4 Pu(Ⅵ)的制备及稳定性 调节Pu(Ⅳ)溶液为0.1mmol/L KMnO4-1mol/L HNO3体系,避光反应24h,用10mL 0.01mol/L NaCl溶液稀释,调节pH=2.5。加入与KMnO4等量的MnCl2沉淀,经0.45μm微孔滤膜过滤后,调节pH=3.0。分析反应前后溶液中钚的价态分布,结果示于图5,由图5可见,反应后溶液中钚价态主要以Pu(Ⅵ)为主,但过滤过程钚活度浓度降低约15%。将制备的Pu(Ⅵ)溶液避光保存,分析不同时间钚价态的变化情况,结果列于表2。由表2可见,在该体系下,Pu(Ⅵ)不能稳定存在,在短时间(5d)时被还原为Pu(Ⅴ),其原因可能为α辐射产生自由基的还原作用[8],继续放置Pu(Ⅴ)又逐渐被氧化为Pu(Ⅵ),可能为溶液中氧气氧化的结果。因此,实验所需Pu(Ⅵ)应新鲜制备。
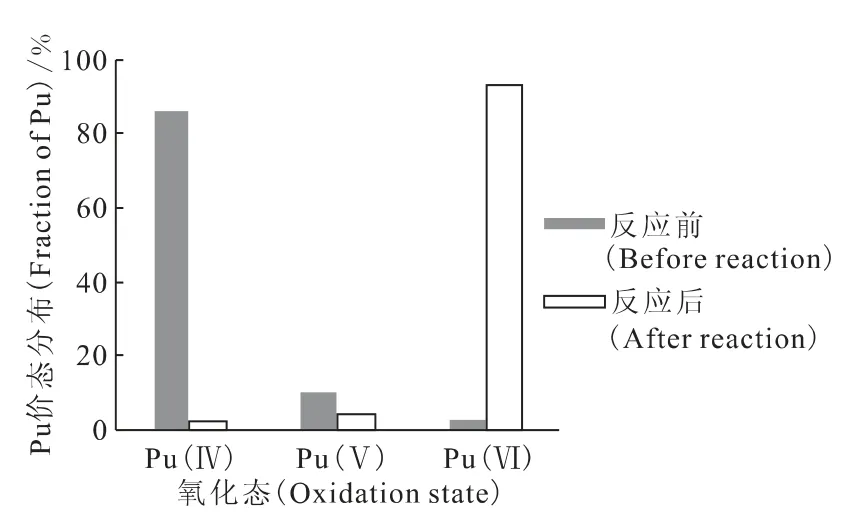
图5 反应前后溶液中钚的价态分布Fig.5 Fraction of Pu oxidation state before and after the reaction

表2 pH=3.0、c(NaCl)=0.01mol/L溶液中238Pu(Ⅵ)的稳定性Table 2 Stabilization of 238Pu(Ⅵ)at pH=3.0,c(NaCl)=0.01mol/L
2.4 Pu(Ⅴ)的制备及稳定性
按确定的实验条件制备浓度分别为1.5×10-10、4.5×10-8、5.7×10-7mol/L的238Pu(Ⅵ)、239Pu(Ⅵ)、242Pu(Ⅵ)溶液,活度浓度均约为20kBq/L,观察Pu(Ⅴ)份额随时间的变化,结果示于图6。由图6可见,放置5d后,浓度较高的六价239Pu和242Pu均被还原为五价,而较低浓度的238Pu仅70%转化为五价,继续放置,五价钚会继续降低并逐步稳定。238Pu、239Pu、242Pu衰变均以α粒子为主,α粒子与水分子作用生成还原性自由基e-(aq)、·H、H2O2,由于自由基的还原作用和Pu(Ⅴ)的热力学稳定性,Pu(Ⅵ)可被还原为Pu(Ⅴ)。实验中3种钚同位素的活度浓度相当,产生自由基的浓度相当[16],放置较长时间(40d)后溶液中五价钚的份额趋于接近。在放置时间较短(5d)时,摩尔浓度较高的239Pu(Ⅵ)、242Pu(Ⅵ)均基本完全转变为五价,这可能与钚的稀溶液化学性质有关[17]。因此,痕量Pu(Ⅴ)溶液的制备需将较高浓度(>10-8mol/L)Pu(Ⅵ)放置5d后制得,然后进行稀释。
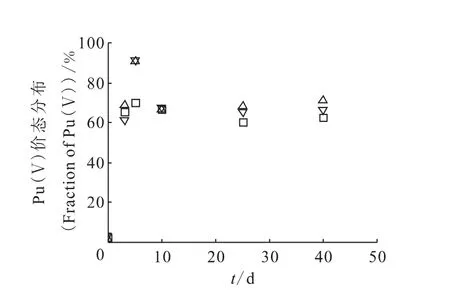
图6 不同钚同位素溶液中五价钚份额随时间变化关系Fig.6 Fraction of Pu(Ⅴ)at different time and isotopes
3 结 论
实验结果表明,采用氧化还原法可成功制得痕量Pu(Ⅳ)、Pu(Ⅴ)、Pu(Ⅵ)溶液,单一价态钚的百分比均在90%以上。Pu(Ⅳ)的制备可在1mol/L HNO3体系反复蒸干制得;Pu(Ⅳ)在0.5~1mol/L HNO3-0.1mmol/L KMnO4溶液体系中反应24h,可制得Pu(Ⅵ)溶液;Pu(Ⅴ)由高浓度(>10-8mol/L)的Pu(Ⅵ)放置5d之后再稀释到痕量水平制得。3种单一价态的钚溶液均应新鲜制备,放置过程中钚的价态分布会发生变化。
[1]Choppin G R.Environmental behavior of actinides[J].Czechoslovak Journal of Physics,2006,56:13-21.
[2]Kersting A B,Efurd D W,Finnegan D L,et al.Migration of plutonium in groundwater at the nevada test site[J].Nature,1999,397:56-59.
[3]Keeney-Kennicutt W L,Geochim M J W.The redox chemistry of Pu(Ⅴ)interaction with commonmineral surfaces in dilute solutions and seawater[J].Cosmochim Acta,1985,49:2577-2588.
[4]Choppin G R.Actinide speciation in the environment[J].Radiochim Acta,2003,91:645-649.
[5]Orlandini K A,Penrose W R,Nelson D M.Pu(Ⅴ)as the stable form of oxidized plutonium in natural waters[J].Mar Chem,1986,18:49-57.
[6]Morse J W,Choppin G R.Laboratory studies of plutonium in marine systems[J].Mar Chem,1989,20:73-89.
[7]Powell B A,Duff M C,Kaplan D I,et al.Plutonium oxidation and subsequent reduction by Mn(Ⅳ)minerals in Yucca mountain tuff[J].Environ Sci Tech,2006,40(11):3508-3514.
[8]Powell B A,Fjeld R A,Kaplan D I,et al.Pu(Ⅴ)adsorption and reduction by synthetic hematite and goethite[J].Environ Sci Tech,2005,39(7):2107-2114.
[9]Romanchuk A Y,Kalmykov S N,Aliev R A.Plutonium sorption onto hematite colloids at femto-and nanomolar concentrations[J].Radiochim Acta,2011,99(3):137-144.
[10]Snow M S,Zhao Pihong,Dai Zurong,et al.Neptunium(Ⅴ)sorption to goethite at attomolar to micromolar concentrations[J].J Colloid Inter Sci,2013,390:176-182.
[11]James D B,Zavarin M,Zhao Pihong,et al.Pu(Ⅴ)and Pu(Ⅳ)sorption to montmorillonite[J].Environ Sci Tech,2013,47:5146-5153.
[12]Cohen D.The absorption spectra of plutonium ions in perchloric acid solutions[J].J Inorg Nucl Chem,1961,18:207-210.
[13]Saito A,Roberts R A,Choppin G R.Preparation of solutions of trace level of plutonium(Ⅴ)[J].Anal Chem,1985,57:390-391.
[14]罗文宗,张文青.钚的分析化学[M].北京:原子能出版社,1991:118-122.
[15]Neu M P,Hoffman D C,Roberts K E,et al.Comparison of chemical extractions and laser photoacoustic spectroscopy for the determination of plutonium in near-neutral carbonate solutions[J].Radiochim Acta,1994,66:265-272.
[16]Hixon A E,Arai Yuji,Powell B A.Examination of the effect of alpha radiolysis on plutonium(Ⅴ)sorption to quartz using multiple plutonium isotopes[J].J Colloid Inter Sci,2013,403:105-112.
[17]Volker N,Marcus A.Solubility of plutonium hydroxides/hydrous oxides under reducing conditions and in the presence of oxygen[J].C R Chimie,2007,10:959-977.
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:40
上海金属(2022年5期)2022-09-26 02:07:28
理化检验-化学分册(2020年5期)2020-06-15 11:36:04
中国测试(2018年4期)2018-05-14 15:33:30
华南农业大学学报(社会科学版)(2015年3期)2016-01-11 11:46:27
西藏科技(2015年1期)2015-09-26 12:09:23
质谱学报(2015年5期)2015-03-01 03:18:47
时代金融(2013年6期)2013-08-15 00:51:28
首都经济贸易大学学报(2013年5期)2013-03-11 18:05:59
植物营养与肥料学报(2011年2期)2011-10-26 03:52:16
