
基于演化学习超网络的微阵列数据分类
2014-12-23 07:14黄萍丽孙开伟
江苏大学学报(自然科学版) 2014年1期
王 进,黄萍丽,孙开伟,蔡 通
(重庆邮电大学计算智能重庆市重点实验室,重庆400065)
癌症治疗是人类难以攻克的难题.DNA微阵列技术的出现为从分子水平研究癌症的发病机理和临床诊断提供了强有力的手段.如何从众多的微阵列数据中挖掘与癌症相关的基因以及基因间的相互作用是当前研究的热点问题[1-3].目前已有多种传统模式识别技术用于分析处理DNA微阵列基因表达谱数据,如支持向量机[4]、人工神经网络[5]等.然而传统模式识别方法的学习结果通常难以解读,对与癌症诊断密切相关的基因高阶关联关系的挖掘也缺乏相应工具.
超网络(hypernetwork)是受生物分子网络启发而建立的一种基于超图(hypergraph)的认知学习模型[6],能够表示模式特征间的高阶关联关系.超网络模型的超边库可以存储训练集的部分信息,同时表示样本的不同属性以及属性和样本类别之间的关联程度.目前超网络已被成功应用于解决多种模式识别与认知问题[7-9].演化学习是超网络模型的关键环节,其目标是从训练集中寻找最佳的特征组合,其学习过程直接影响超网络的分类效果[9].传统超网络的学习方法主要包括2种,一种是梯度下降法[7]:在演化过程中,根据某种策略对初始化后的超边权值进行调整.该学习方法简单且易操作,其主要局限在于超网络对超边空间的搜索具有局部性.如果在初始化过程中超网络没有提取出有用的特征组合,往往无法取得较好的分类效果.另一种常用的学习方法是超边替代法[8-9],它采用随机搜索的方式寻找超边,虽然可以扩大搜索空间,但随机搜索效率较低且不稳定.
针对传统超网络学习算法的局限,文中将遗传算法与超网络模型相结合,提出一种基于遗传算法的演化超网络模型(genetic algorithm-based evolutionary hypernetworks,GA-HN).采用遗传算法中的锦标赛选择算子对超边进行选择,并通过交叉和变异算子产生新的超边后代.
1 超网络模型
1.1 超网络定义
超网络是超图的推广,在介绍超网络模型之前先给出超图的数学定义.超图G是由超边组成的无向图,超边可以连接任意多个顶点,即G=(V,E)[9].其中V={v1,v2,…,vn}是包含n个顶点的集合,E={e1,e2,…,em}是包含m条超边的集合.E中每个元素ei={vi1,vi2,…,vik}(k≥1)是一条连接k个顶点的超边.
超网络模型对超图的每条超边赋予权值和类别标识,可以用一个三元组H表示,H=(V,E,W).其中V是特征变量集合,E={e1,e2,…,em}为超网络的超边集合.超边ei={vi1,vi2,…,vik,yi}由k个特征及类别标识yi组成,k称为超边的阶数(1≤k≤n),n表示特征空间V的大小.W={w1,w2,…,wm}是超边的权值集合.图1是一个包含7个特征4条超边的超网络,图中超边线条的粗细代表了超边权值的大小.

图1 一个包含7个特征4条超边的超网络
1.2 超网络分类器
根据超网络定义,可以把超网络看作一个分类器.超网络的超边作为决策规则,对输入模式的类型进行判断.当输入一个待识别样本X,超网络分类器利用所有超边共同做出决策,输出该模式所对应的类别Y.
根据输入训练集D,对其中每个样本的特征进行多次随机采样,建立一个初始化超边库,如图2所示.(超网络初始化及演化学习过程详见第2.2节).D具有以下形式:
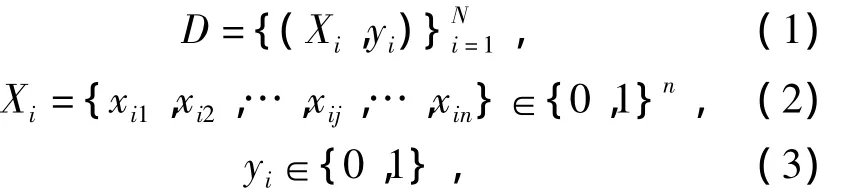
式中:Xi为第i个训练样本;yi为Xi的类别标识;N为训练集样本总数;xij为第i个样本的第j维特征的表达值;n表示特征的空间大小.超网络分类器H(X)可以表示输入样本X与输出类别Y∈{0,1}的联合概率P(X,Y).该联合概率可以通过对超边进行点估计近似得到:
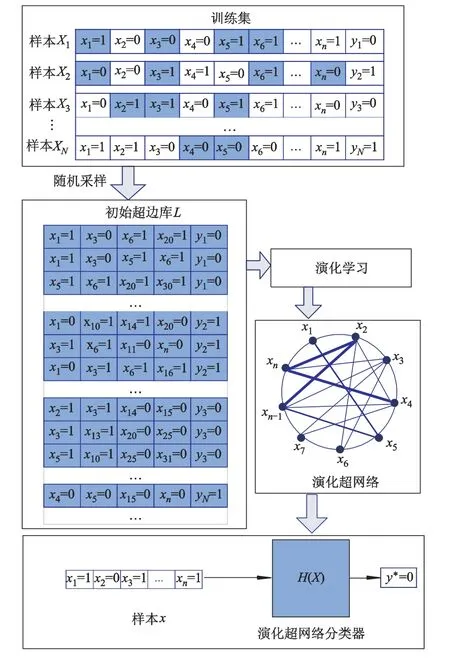
图2 超网络分类器的建立过程

式中:|L|为超边库的规模;MY表示与输入样本X正确匹配且类别为Y的超边数;(xi1,xi2,…,xik,Y)为第i条超边的决策规则.当第i条超边和样本X正确匹配时,(xi1,xi2,…,xik,Y)为 1,否则为 0.
超边与样本正确匹配定义如下:超边包含的顶点特征值与样本对应的特征值相等,且超边的类别标识与样本的类别相同.例如:对于一个k(k=3)阶超网络模型,输入样本X={x1=1,x2=0,x3=0,x4=1,y=0}.若一条超边ei的顶点为xi1=1,xi3=0,xi4=1,则称超边ei与样本X匹配.若ei与样本X匹配且类别相等,则称超边ei与样本X匹配正确,反之匹配错误.
在分类应用中,超网络首先计算输入X属于每个类别的条件概率,然后取条件概率最大的类别作为分类结果:


也就是说,给定一个输入样本X,超网络将所有超边与X进行匹配,并将匹配的超边按类别进行划分.取匹配数量最多的一类超边类别作为该输入样本X的分类结果y*=H(X).若无任何超边与X匹配或者匹配的两类超边数相等,即样本被判为任何一类的概率相等,超网络将该样本判为类别1.
超网络分类器的分类过程如下:
1)输入待分类样本X;
2)根据以下步骤对输入样本X进行分类:
①将X与L中所有超边进行匹配运算,并将与X匹配的超边放入集合M中.
②根据超边的类别标识,对集合M中的超边进行划分:类别为0的超边归类到M0中,将类别为1的超边归类到M1中.
式中:P(X)≈|M|/|L|表示输入样本X被超网络学习记忆的概率,该值通过计算所有与输入样本X匹配的超边数|M|与超边总数的比值得出.由式(4)和式(5)得
在这一过程中,超网络利用超边数量众多这一特性,使分类结果具有较强的鲁棒性.
2 基于遗传算法的超网络演化学习
2.1 超网络的遗传算法编解码
演化学习以超边的顶点组合为对象,将特征变量空间进行二进制遗传算法编码.初始化后,超边所包含的特征变量用二进制编码表示,得到的二进制串作为遗传算法染色体.在整个演化学习过程中,超边库中的超边以染色体编码串形式存在,超边顶点对应的属性值被隐藏.最后需根据训练集对演化后的超边进行解码,返回超边顶点对应的属性值.
当特征空间大小为n时,每个特征需用t位二进制数进行编码表示,其中2t≥n.具有k(k表示超边的阶数)个顶点的超边需要长度为kt+1的染色体来表示,其中最后一位用于表示超边的类别标识.遗传算法在超网络中的编解码过程如图3所示.
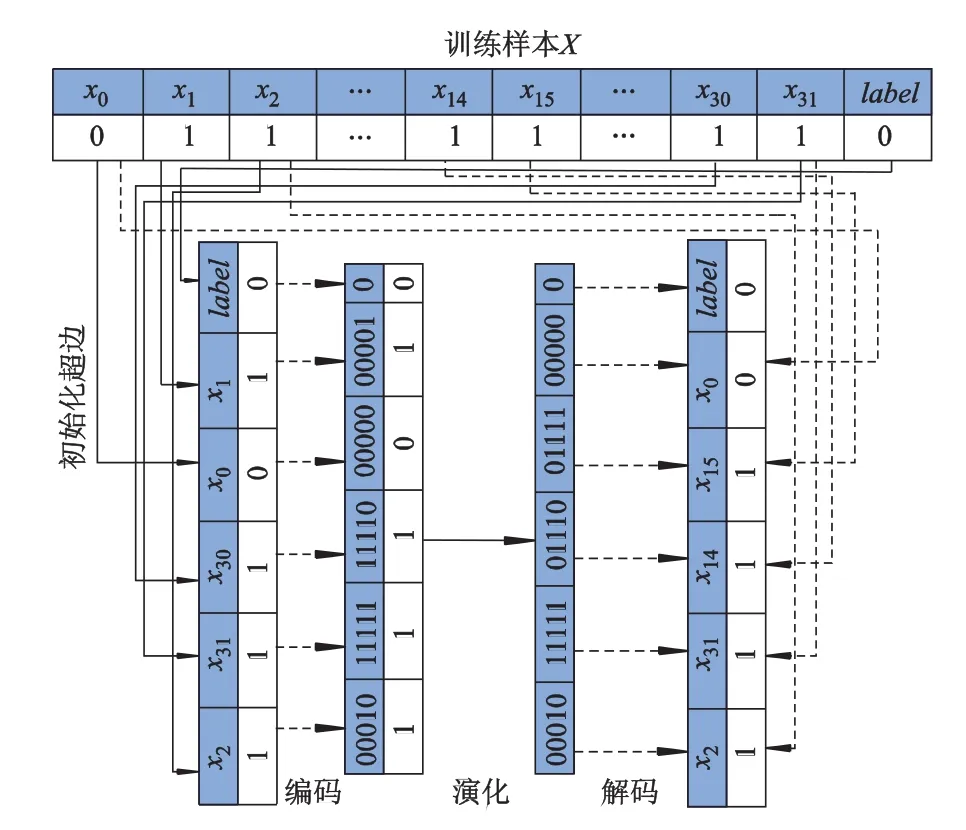
图3 超边的遗传算法编解码过程
2.2 超网络演化学习
学习算法的性能直接影响超网络的学习速度和识别率.文中采用并行遗传算法对超网络进行演化,演化流程如图4所示.

图4 超网络演化学习流程图
演化的具体步骤如下.
1)初始化超边
初始化超网络的子种群.对于训练集D中的每个样本xi,从n个特征中随机抽取k个特征,组成一条k阶超边li,初始化超边的适应值fit(li)=0.对每个样本生成T条超边,最终构成具有|D|×T条超边的超边库L,其中,|D|为训练集样本总数.将初始化后的超边库L进行编码,并随机分成M个子种群,每个子种群包含m条超边(每条超边对应子种群中的一个个体).由于各个子种群都执行相似的演化学习过程,下面以其中一个子种群为例,说明其演化学习过程.
2)超网络分类
根据1.2节中介绍的分类过程,由每条超边对训练集进行分类.
3)个体适应值的计算
个体适应值表征超边的分类能力.根据超边对训练集的分类结果,计算个体适应值,以评估超边的分类性能.当超边与训练集某个样本正确匹配时,该超边的准确值#c加1;否则,超边的错误值#w加1.个体适应值定义为

式中:α=100为错误预期值;β=1为正确预期值.
3)锦标赛选择
每次从子种群中随机抽取a个个体进行适应值对比(文中取a=2),其中适应值最大的个体将被遗传到下一代;重复以上过程m次,就可得到下一代种群的m个个体.
4)迁移
对经锦标赛选择后的超边按适应值大小进行排序,将适应值最大的q条超边迁移到左邻接子种群,代替其中适应值最低的q条超边,保证优良个体及时扩展到邻接子种群中.
5)交叉操作
从步骤4)得到的子种群中,以一定的交叉概率pc随机选择类别相同的2条超边,在超边的某一顶点处进行单点交叉操作,产生2个新的个体.
6)变异操作
采用基本位变异法,按一定的变异概率pm随机指定某个体编码串中的一位基因进行变异操作.若被指定进行变异操作的二进制位为0,则将其变为1;反之,若为1,则将其变为0.
7)返回步骤2),直到超网络完成预定的演化代数epoch.
3 试验结果分析
为了验证GA-HN的分类性能,文中采用急性白血病、肺癌、前列腺3个数据集作为试验用例.急性白血病数据集来源于麻省理工学院采集的72个不同病人的白血病样本,每个样本包含6 817个基因,分为ALL和AML 2种类型.其中38个样本(27个ALL,11个AML)作为训练集;另外独立的34个样本(20个ALL,14个AML)作为测试集.肺癌数据集包含恶性胸膜间皮瘤(malignant pleural mesothelioma,MPM)和肺腺瘤(lung adenocarcinoma,ADCA)两种类型,共181个样本,每个样本12 533个基因.其中32个样本(16个MPM,16个ADCA)作为训练集;另外独立的149个样本(15个MPM,134个ADCA)作为测试集.前列腺数据集包含102个训练集样本(52个癌细胞样本和50个正常样本)和34个测试集样本(包含25个癌细胞样本和9个正常样本),样本基因数为12 600.通过对3个癌症数据集进行信噪比特征选择[10],文中选取样本中的32个基因进行分类试验.表1给出了GA-HN在急性白血病、肺癌和前列腺3个数据下遗传算法的相关参数设定.文中所有平均识别率都是基于对测试集100次测试的结果.

表1 GA-HN参数设定
在基于遗传算法的超网络演化学习方法中,超边阶数的设定对超网络的分类结果有直接影响.如图5所示,针对不同的数据集,超网络的平均识别率都随阶数的变化而变化.超边阶数过低或过高,分类的效果都不理想.这是由于当超边的阶数过低时,超边中所包含的特征信息过少,遗漏了某些关键属性;而当阶数过高时,超边包含的噪声和冗余信息过多,也会降低超网络的分类效果.由图可得超网络阶数分别设为12,5,11时,超网络分类器对急性白血病、肺癌和前列腺数据集的识别准确率最高.

图5 超网络识别率随阶数的变化
表2给出了GA-HN对3种数据集的试验结果.由表2可以看出,在给定参数条件下,GA-HN具有较高的分类正确率;在对急性白血病、肺癌、前列腺3个数据集100次的分类试验中,GA-HN分类准确率的标准差非常小,分别为2.84%,0.20%,2.90%,说明GA-HN系统稳定性良好.这是由于超网络利用众多的超边共同做出决策,因此分类器表现出较好的稳定鲁棒性.
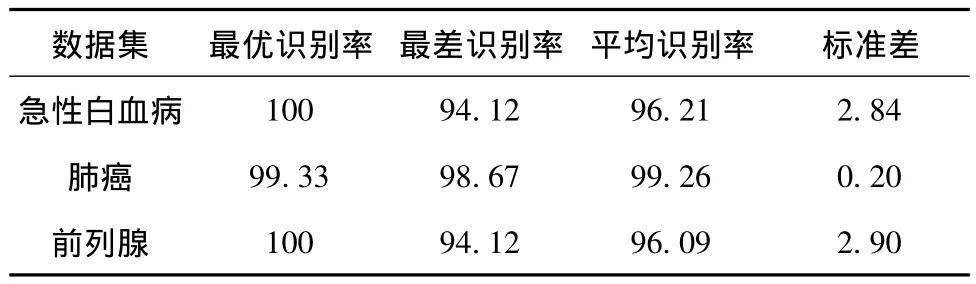
表2 GA-HN对3种癌症数据的识别率 %
超网络演化学习策略的选择对系统分类性能也有较大影响.表3给出了GA-HN与基于超边替代策略的超网络(random feature subset hypernetworks,RFS-HN)以及基于梯度下降策略的超网络(gradient descent-based hypernetworks,GD-HN)在对3个癌症数据集下的对比试验结果.由表3可见,与传统的RFS-HN和GD-HN相比,文中所提出的方法具有更高的平均识别率.

表3 基于不同演化学习策略的超网络平均识别率对比%
表4,5,6给出了GA-HN与其他传统模式识别方法包括神经网络(neutral network,NN)、遗传规划(genetic program,GP)、演化硬件(evolvable hardware,EHW)、基于 Bagging的决策树 C4.5 算法(Bagging-C4.5)、组合GAM和CCM分类算法(ensemble algorithm based on GCM and CCM,EAGC)对3个癌症数据集的识别结果对比,可见对于急性白血病和前列腺数据集,GA-HN的识别率均在该领域传统方法的平均水平之上;而对于肺癌数据集,GA-HN在识别率上相对 Bagging-C4.5有较大提升.
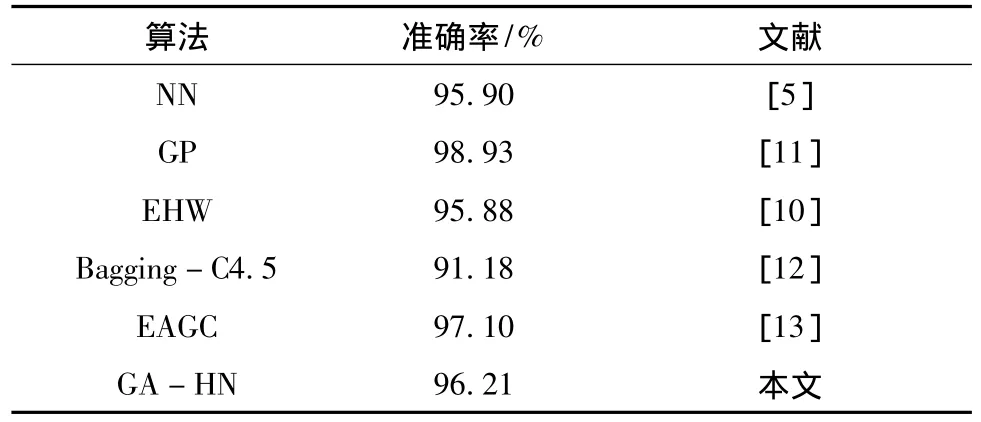
表4 不同分型方法对急性白血病数据集的识别率对比

表5 不同分型方法对肺癌数据集的识别率对比
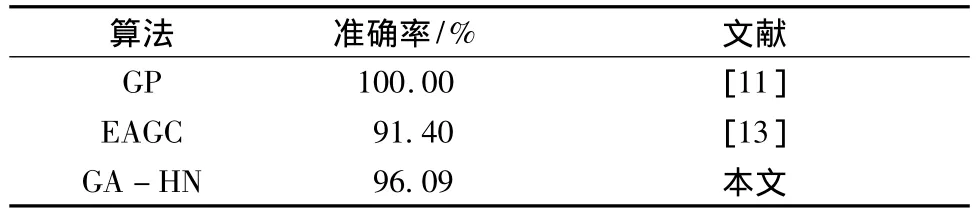
表6 不同分型方法对前列腺数据集的识别率对比
超边的适应值可以表征超网络的识别性能.图6是GA-HN和RFS-HN在演化过程中超边库平均适应值的变化曲线.

图6 基于不同学习策略的超网络演化过程
图6a,b,c分别为3类数据集的试验结果,由图6可见,超网络在演化学习过程中,超边库的平均适应值均呈上升趋势.说明超边可以对其所覆盖的大部分训练集数据进行正确分类,超边的分类能力随学习进程不断增强.RFS-HN采用超边替代的策略进行演化学习,在超边替代过程中,新的超边通过随机选择产生.该方式虽然扩大了超边的搜索空间,但具有不确定性,因此其超边库适应值的上升速度明显低于文中提出的GA-HN.由图6还可以看出,经过10代基于遗传算法的演化学习,超网络超边库的平均适应值趋于平稳,说明超网络趋于收敛.
通过演化学习,超网络能够有效挖掘与模式识别相关的特征组合.表7给出了GA-HN发现的与肺癌相关的前10个具有较高适应值的5阶基因组合.包含这些基因组合及相应特征取值的超边可以有效识别相应的样本类型,使试验结果具有良好的可读性.例如,从表7中可以看出,当超边包含特征组合{35276-at,291-s-at,39409-at,2047-s-at,266-s-at},且对应取值为0,0,1,0,0时,超边对16个ADCA类型的训练样本分类完全正确.意味着当样本中包含上述基因组合及相应属性取值时,超网络可以以很大的概率将其判定为ADCA类别.

表7 与肺癌相关的高阶基因组合

续表
4 结论
文中将遗传算法引入超网络的演化学习过程,提出了一种基于遗传算法的演化学习超网络模型,得到如下结论:
1)基于遗传算法演化学习的超网络分类器具有较高的分类准确率,并且其演化学习结果易于理解.
2)超网络分类器利用数量众多的超边进行分类,其结果具有更高的可靠性和稳定性.
3)演化超网络能有效挖掘癌症基因对间的相互作用.因此,超网络模型可作为生物信息处理,尤其是疾病诊断方面的有效数据挖掘工具.
References)
[1]于化龙,顾国昌,赵 靖,等.基于DNA微阵列数据的癌症分类问题研究进展[J].计算机科学,2010,37(10):16-22.Yu Hualong,Gu Guochang,Zhao Jing,et al.State of the art on cancer classification problems based on DNA microarray data[J].Computer Science,2010,37(10):16-22.(in Chinese)
[2]Dagliyan O,Uney-Yuksektepe F,Kavakli I H,et al.Optimization based tumor classification from microarray gene expression data[J].PLoS One,2011,6(2):e14579.
[3]王 年,庄振华,范益政,等.癌症基因分类的Laplace谱方法[J].电子学报,2011,39(7):1594-1597.Wang Nian,Zhuang Zhenhua,Fan Yizheng,et al.Classification of tumor gene expression data based on laplacian spectra of graphs[J].Acta Electronica Sinica,2011,39(7):1594-1597.(in Chinese)
[4]Nanni L,Brahnam S,Lumini A.Combining multiple approaches for gene microarray classification[J].Bioinformatics,2012,28(8):1151-1157.
[5]Cho S B,Won H H.Cancer classification using ensemble of neural networks with multiple significant gene subsets[J].Applied Intelligence,2007,26(3):243-250.
[6]Lee J H,Lee B,Kim J S,et al.A molecular evolutionary algorithm for learning hypernetworks on simulated DNA computers[C]∥Proceedings of2011IEEE Congress of Evolutionary Computation.New Orleans:IEEE Computer Society,2011:2735-2742.
[7]Bootkrajang J,Kim S,Zhang B T.Evolutionary hypernetwork classifiers for protein-protein interaction sentence filtering[C]∥Proceedings of Genetic and Evolutionary Computation Conference.New York:ACM,2009:185-194.
[8]王 进,金理雄,孙开伟.基于演化超网络的中文文本分类方法[J].江苏大学学报:自然科学版,2013,34(2):196-201.Wang Jin,Jin Lixiong,Sun Kaiwei.Chinese text categorization based on evolutionary hypernetwork[J].Journal of Jiangsu University:Natural Science Edition,2013,34(2):196-201.(in Chinese)
[9]王 进,孙开伟,李钟浩.超网络道路限速标志识别[J].小型微型计算机系统,2012,33(12):2709-2714.Wang Jin,Sun Kaiwei,Lee Chong-ho.Hypernetworks for road speed limit sign recognition[J].Journal of Chinese Computer Systems,2012,33(12):2709-2714.(in Chinese)
[10]王 进,陈 文,李钟浩.用于癌症分子分型的虚拟可重构结构演化硬件[J].华中科技大学学报:自然科学版,2012,40(4):23-28.Wang Jin,Chen Wen,Lee Chong-ho.Virtual reconfigurable architecture-based evolvable hardware for molecular classification of cancers[J].Journal of Huazhong U-niversity of Science and Technology:Natural Science Edition,2012,40(4):23-28.(in Chinese)
[11]Liu Kunhong,Xu Chungui.A genetic programmingbased approach to the classification of multiclass microarray datasets[J].Bioinformatics,2009,25(3):331-337.
[12]Tan A C,Gilbert D.Ensemble machine learning on gene expression data for cancer classification[J].Applied Bioinformatics,2003,2(3 suppl):75-83.
[13]卢新国,林亚平,骆嘉伟,等.癌症识别中一种基于组合GCM和CCM 的分类算法[J].软件学报,2010,21(11):2838-2851.Lu Xinguo,Lin Yaping,Luo Jiawei,et al.Classification algorithm combined GCM with CCM in cancer recognition[J].Journal of Software,2010,21(11):2838-2851.(in Chinese)
猜你喜欢
计算机工程(2020年3期)2020-03-19
中国听力语言康复科学杂志(2019年3期)2019-06-24
中国交通信息化(2018年3期)2018-06-13
光学精密工程(2016年4期)2016-11-07
光学精密工程(2016年3期)2016-11-07
中国交通信息化(2016年2期)2016-06-06
新校长(2016年8期)2016-01-10
商事法论集(2014年1期)2014-06-27
电测与仪表(2014年15期)2014-04-04
中国中医药现代远程教育(2014年16期)2014-03-01
