
氧化法制备1,2-苯并异噻唑啉-3-酮
2014-12-23 01:00张可青任蕊杨俊伟李程碑
应用化工 2014年9期
张可青,任蕊,杨俊伟,李程碑
(陕西省石油化工研究设计院,陕西 西安 710054)
异噻唑啉酮是一类重要的工业杀菌剂,具有抗菌能力强、应用剂量小、相容性好、药效持续时间长、对环境安全以及抗菌谱宽广等优良性能[1],在工业、农业、医药等行业得到了广泛应用。在诸多异噻唑啉酮化合物中,1,2-苯并异噻唑啉-3-酮(BIT)因具有优异的耐温性和耐酸碱能力,在防腐杀菌领域受到越来越多的关注[2],已经在诸多常规防腐产品难以发挥效果的特殊工业领域中显示出了独特的优势。
长期以来,BIT 的合成方法多采用卤素氧化法[3],该法需使用氯气/液溴等剧毒、强腐蚀原料,使用极其不便,且过量的卤素吸收不完全,极易造成二次污染,已经难以达到目前的环保要求。如何在不使用卤素的前提下寻找产率高、价格低廉且工艺简单的BIT 新型生产工艺,成为了防腐剂生产领域的热门问题。
本文提出了一种简便环保的合成BIT 的方法,即以二硫化二苯甲酸为起始原料,通过酰氯化、酰胺化反应,然后采用双氧水氧化法制备得到BIT;产物结构通过红外光谱加以确认,反应总收率67.7%。该法以双氧水氧化过程代替传统工艺中卤素的氧化过程,合成过程清洁环保,成本低廉,且产率更高,产品纯度更好,对于BIT 的大规模工业生产具有很大的意义。
1 实验部分
1.1 试剂与仪器
二硫化二苯甲酸、氢氧化钠、氯化亚砜、吡啶、丙酮、甲苯、氨水(25%)、双氧水(30%)、乙醇均为化学纯;浓盐酸,工业品。
WRS-2 型熔点测定仪;VX-70 型红外吸收光谱仪。
1.2 实验方法
合成过程按照如下反应方程式分步进行:
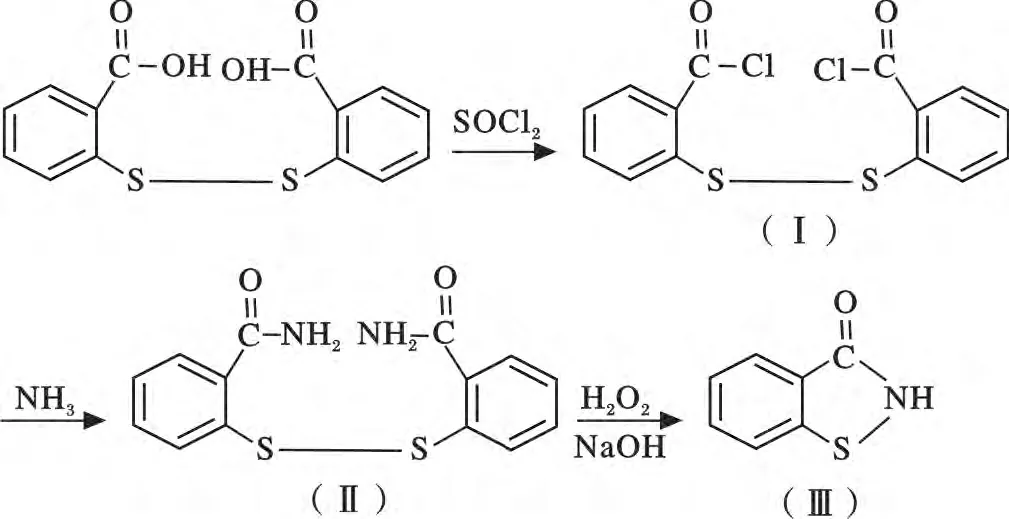
1.2.1 2,2-二硫化二苯甲酰氯的合成( Ⅰ) 在带有回流和尾气吸收装置的500 mL 四口烧瓶中,加入30.6 g(0.1 mol)二硫化二苯甲酸、100 mL 甲苯和8 mL吡啶,开动搅拌,在室温下缓慢滴加47. 6 g(0.4 mol)氯化亚砜,滴完后将反应温度升至60 ~70 ℃,保温反应1 h,停止加热,减压蒸馏回收过量的氯化亚砜(温度35 ~40 ℃),得棕黄色悬浮液。抽滤,用丙酮洗涤,在50 ℃真空烘箱中干燥8 h,即得黄色2,2-二硫化二苯甲酰氯粉末,熔点154 ~157 ℃(文献值155 ~159 ℃),反应收率93.3%。
1.2.2 2,2-二硫化二苯甲酰胺的合成( Ⅱ) 在500 mL 三口烧瓶中加入100 mL(约1.5 mol)氨水和50 mL 甲苯,开动搅拌,控制温度在30 ℃以下,分次加入34.3 g(0.1 mol)(Ⅰ),在室温下反应1 h。抽滤,以水洗涤数次,在50 ℃真空干燥8 h,即得淡黄色2,2-二硫化二苯甲酰胺固体,熔点197 ~202 ℃,与文献相符,反应收率95.4%。
1.2. 3 BIT 的合成( Ⅲ)在带有回流装置的500 mL四口瓶中加入15.2 g(0.05 mol)(Ⅱ)和约100 mL 氢氧化钠水溶液(含氢氧化钠8. 0 g/0.2 mol),升温至60 ℃,保持该温度,搅拌下在2 h内缓慢滴加20.4 g 10% 双氧水溶液(0. 06 mol H2O2),滴加完后降温至50 ℃,继续反应1 h。冷却至室温,用稀盐酸调节pH 为4 ~5,放入冰箱冷冻10 min。过滤,用水洗涤至中性,再用少量乙醇洗涤,即得白色BIT 固体,熔点156 ~158 ℃,与文献报道相符合,反应收率76. 1%。IR(γ/cm-1):3 056(Ar—H),2 909,2 665(—CH3,—CH2—),1 638( C O ),1 443,1 461(苯环骨架),1 316(N—S),1 147(C—N),742(苯环上邻二取代),606(C—S)。
2 结果与讨论
由于本文合成过程的前两步,即羧酸酰氯化和酰胺化过程均属经典反应,在这里不作探讨。本文重点对双氧水氧化成环的关键一步进行讨论。
2.1 氧化反应及成环机理
异噻唑啉酮杂环的闭合是通过—S—S— 键的断裂和—N—S— 键的形成来完成的,—S—S— 键的断裂为自由基取代历程[4],而—N—S— 键的形成为亲核取代历程[5],总反应历程推测为:双氧水在碱性条件下生成HO·自由基,进攻—S—S— 键使其断裂,生成RS—OH 和RS·自由基,RS·自由基与双氧水反应,再次生成RS—OH 和HO·自由基,从而使得自由基取代反应持续进行。在碱性条件下,酰胺基氮原子上的孤对电子进攻带有微量正电荷的RS—OH,OH-离去,与氮原子上脱去的H+结合生成H2O,同时N—S 键形成,从而形成较为稳定的苯并异噻唑啉酮五元环。
—S—S— 键断裂反应:

—N—S— 键生成反应:

2.2 双氧水滴加时间对反应的影响
本反应中双氧水分解所得的HO·自由基可将—S—S— 键氧化断裂,但如果体系中双氧水浓度过高,自由基产生的速度快于环合反应,则多余的自由基易于将断键生产的RS— 进一步氧化为硫醇的氧化副产物和磺酰化合物[6]。为避免单位时间内反应体系中氧化剂浓度太大而导致产物过氧化,双氧水需在整个氧化过程内缓慢加入。在不同反应时间下进行了氧化反应,实验结果列于表1。
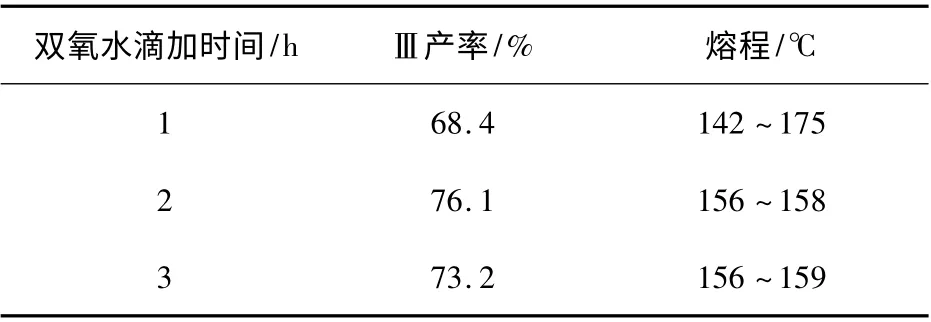
表1 双氧水滴加时间对反应的影响Table 1 Effect of drop time of hydrogen peroxide on the reaction
由表1 可知,双氧水在2 h 内滴加完时,产率最高,且熔程仅为2 ℃,表明产物纯度很好。滴加时间缩短为1 h 时,产率降低,熔程延长,其中一部分产物的熔点在170 ℃以上,表明反应没有进行完全。滴加时间延长为3 h 后,产率低于滴加时间2 h 所得产物,且产物纯度较低,表明反应时间延长导致过氧化反应的发生。上述现象均说明双氧水滴加时间对于氧化反应的影响很大,时间太短,反应不能进行完全,而时间太长,反应易于发生过氧化副反应。因此双氧水滴加时间以2 h 为最佳。
2.3 氧化剂用量对反应的影响
由于氧化剂过量可引起产物过氧化,因此氧化剂与酰胺的摩尔比不宜过高。表2 为不同氧化剂用量所得反应产率。

表2 氧化剂用量对反应的影响Table 2 Effect of amount of oxidant on the reaction
由表2 可知,氧化剂双氧水与酰胺的摩尔比为1∶1.1 和1∶1.2 时,反应产率最高;此后,随着双氧水与酰胺的摩尔比逐渐增大,反应产率逐渐降低,当双氧水与酰胺的摩尔比为1∶2 和1∶3 时,氧化剂大幅过量,产物大量过氧化,反应产率急剧下降。考虑到双氧水在反应前可能出现少量分解的情况,双氧水与酰胺的最佳摩尔比应为1∶1.2。
2.4 碱性强弱对氧化反应的影响
为研究反应体系碱性强弱对反应产率的影响,在pH 为8 ~14 的条件下进行了合成反应。实验表明,当反应体系pH 低于9 后,二硫化二苯甲酰胺在反应过程中未能完全溶解,而反应产率也明显降低,这可能是因为未溶解的酰胺难以发生氧化反应。熔点测定表明,大部分产物在标准熔点157 ℃左右熔解,但少量产物在180 ℃以上仍不熔,表明产物中有未反应的酰胺,证实了上述推论。当反应体系pH值在9 ~14 时,酰胺均可完全溶解,反应产率较高。
2.5 质子催化剂对氧化反应的催化作用
在传统的卤素氧化法合成BIT 的过程中,常使用碱性催化剂来催化闭环反应[7],包括氢氧化钠、氢氧化钾等无机碱和吡啶、三乙胺等有机碱。另据报道[8],在双硫键的氧化断裂反应中,含N、P、S 的质子亲核试剂可催化氧化断键反应,使双硫键断裂反应的时间明显缩短,使用较多的此类亲核试剂包括三乙胺、氨水、酰胺、硫脲等。为研究上述催化剂对本反应的影响,选择三乙胺为催化剂进行了合成反应,为避免氧化时间过长导致过氧化发生,加入催化剂后氧化反应时间缩短为2 h,反应结果见表3。
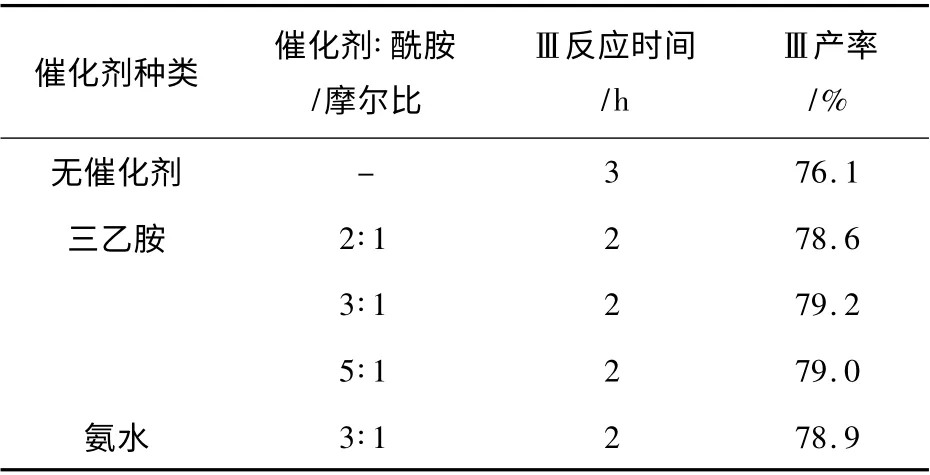
表3 质子催化剂对反应的影响Table 3 Effect of catalyst on the reaction
由表3 可知,在碱性强弱和反应温度等条件相同的前提下,加入催化剂三乙胺,使反应时间明显缩短,收率提高。在加入与酰胺摩尔比分别为2 ∶1,3∶1和5 ∶1 的三乙胺后,反应2 h 的收率分别为78.6%,79.2%和79.0%,均高于未加催化剂反应3 h 后收率,表明三乙胺对该反应具有较好的催化作用。三乙胺对于双氧水氧化制备BIT 反应的催化作用分为两步,分别为催化双硫键断裂的自由基反应和催化闭环亲核反应,由于自由基反应为慢反应,而亲核反应为快反应,因此三乙胺对双硫键断裂过程的催化作用为反应加快的主要因素。
此外,由于含有N、P、S 的碱性化合物均可以作为双氧水氧化反应的催化剂,本文还尝试了以氨水作为催化剂进行反应。当反应体系加入与酰胺摩尔比1∶3 的氨水并反应2 h 后,产率达到78.9%,表明氨水对双氧水氧化反应具有较好的催化作用。由于氨水是合成酰胺的原料,且用量大幅过量,因此可考虑将1.2.2 节中未加提纯的酰胺反应产物直接与双氧水进行氧化反应,既可简化合成步骤,又可将过量氨水直接作为催化剂,对于工业生产具有一定意义。
3 结论
(1)以二硫化二苯甲酸为起始原料,通过酰氯化、酰胺化制备得到二硫化二苯甲酰胺,然后在碱性条件下与双氧水进行氧化反应,经盐酸中和,即得1,2-苯并异噻唑啉-3-酮。产品结构经熔点测定和红外光谱得到确认。
(2)双氧水氧化成环反应由双硫键断裂的自由基反应和环合亲核反应两步组成,氧化反应受双氧水滴加时间、用量、体系pH 值以及催化剂的影响。在酰胺与双氧水摩尔比1∶1.2,pH 值9 ~14,加入与酰胺摩尔比3∶1 的三乙胺为催化剂的条件下,氧化时间2 h 所得产率最高,总收率为67.7%。
[1] 李淑琏,李志勇.高效工业杀菌防霉剂异噻唑啉酮类化合物[J].广州化工,1994(3):10-13.
[2] 张瀚,张书成,陈愉江,等.杀菌防腐剂1,2-苯并异噻唑啉-3-酮的合成及应用[J]. 精细化工,1990,7(2):61-64.
[3] 倪越,严万春,耿金懿,等.新工业防霉杀菌剂1,2-苯并异噻唑啉-3-酮的研制[J]. 精细与专用化学品,2004,12(20):21-22.
[4] 密尔顿.有机反应机理的基本原理[M].王建华,译.上海:上海科学技术出版社,1984.
[5] 方永勤,张雁. N-甲基-1,2-苯并异噻唑啉-3-酮的合成研究[J].化学世界,2009,50(6):361-363.
[6] B.G.科克斯,T.格雷.制备苯并异噻唑啉-3-酮的方法:CN,1246114A[P].2000-03-01.
[7] 李育麟,李淑琏,李志勇.N-正辛基-1,2-苯并异噻唑啉-3-酮的合成研究[J].华南师范大学学报:自然科学版,1997(4):53-58.
[8] Tonne Peter,Jaedicke Hagen. Preparation of 1,2-Benzisothiazolinone:Canadian,P3500577.7,[P].1985-01-10.
猜你喜欢
食品科学技术学报(2022年6期)2022-12-15
化工学报(2022年10期)2022-11-13
化工与医药工程(2022年3期)2022-08-08
河南化工(2020年11期)2020-12-10
广州化工(2020年5期)2020-04-01
氯碱工业(2020年7期)2020-03-02
中国抗生素杂志(2019年9期)2019-10-10
浙江化工(2018年3期)2018-04-19
中国资源综合利用(2016年6期)2016-01-22
郑州大学学报(工学版)(2015年1期)2015-03-24
