
炔硒醚的磷氢化反应*
2014-12-19 05:26:20喻爱和邱仁华许新华
湖南大学学报(自然科学版) 2014年7期
喻爱和,邱仁华,许新华†
(1.湖南大学 化学化工学院,湖南 长沙 410082;2.湖南机电职业技术学院,湖南 长沙 410151)
硒原子具有空的4d轨道,对碳负离子有很好的稳定作用,芳硒基基团在有机合成中常作为保护基团.例如,用witting反应制备炔烃是一种重要的合成方法(Scheme 1)[1].然而为了形成稳定的磷叶立德中间体,R1必须是吸电子基如酯基,氰基等.而且这种方法具有局限性,不能制备端炔.
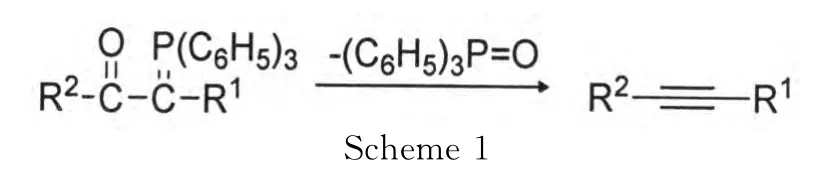
为了扩大这一方法的适用范围,改进的方法就是在与磷相连的碳原子上引进芳硒基,通过消除生成炔硒醚,炔硒醚脱去芳硒基就可以得到端炔(Scheme 2)[1]:

关于炔硒醚脱保护,已经有一些文献报道,如使用正丁基锂或者间氯过氧苯甲酸脱保护[2];使用Cp2TiCl2/i-tBuMgBr体系脱保护[3];使用三丁基锡烷与偶氮二异丁氰使炔硒醚脱保护等[4].这些方法使用的正丁基锂及格式试剂对水十分敏感,需要严格的无水操作;间氯过氧苯甲酸及偶氮二异丁氰是易爆的危险物质.因此,发展简便的去硒化方法,对于端炔的制备具有很大意义.
炔硒醚是合成转化的重要中间体[5-17].通过与亲电试剂或亲核试剂加成,可以得到双官能团烯烃.氢氧化铯是无机超强碱,它能与弱酸性物质反应形成强亲核性的阴离子[18-19].P-H 键是极性键,氢表现出一定酸性,它应能与氢氧化铯反应形成亲核性磷负离子.因此,设想氢氧化铯催化O,O-二烷基亚膦酸酯与炔硒醚进行加成,以期制备含磷和硒的新型的双官能团烯烃.但是,实验表明,氢氧化铯催化下,亚磷酸酯并不能对炔硒醚进行加成,而是发生炔硒醚的去硒化反应.本文报道这一研究结果.
1 实验部分
1.1 仪器与试剂
31P NMR(TMS为内标),1H NMR(TMS为内标),13C NMR(以TMS 为内标)用INOVA-400型仪测定,质谱由HP5989A 测定.溶剂DMF 未经除水处理,硅胶为青岛海洋化工厂产品,氢氧化铯从Aldrich 公司购买.
1.2 实验方法
在室温及氮气下,将1.0 mmol的亚磷酸二乙酯与0.2mmol氢氧化铯加入到5.0mL DMF中搅拌0.5h,然后加入1.0 mmol 炔硒醚继续搅拌,TLC追踪反应进程,待炔硒醚消耗完,停止反应,加入20mL水,用乙酸乙酯(15mL×3)萃取,合并有机相,有机相用水洗(20mL×2),无水硫酸钠干燥.柱层析纯化,先用纯石油醚做洗脱机将端炔冲下来,再用石油醚/乙酸乙酯(V∶V=1∶1)做洗脱机将磷酸硒酯分离出来,即得产物2a~2f及3a~3d.
乙酰氧基丙炔(2a)[20]:Oil;1H NMR(400MHz,CDCl3)4.63(s,2H),2.45(s,1H),2.12(s,3H);13C NMR (400 MHz,CDCl3)170.0,78.2,76.5,55.6,19.5;Ms(m/z):98(M+).
苯氧基丙炔(2b)[21]:Oil;1H NMR (400 MHz,CDCl3)7.33-7.29(t,J=3.1Hz,2H),7.00-6.98(m,3H),4.70(d,J=2.3 Hz,2H),2.53-2.52 (t,J=2.4 Hz,1H);13C NMR(400 MHz,CDCl3)157.7,129.1,121.3,114.8,78.5,76.1,56.4;Ms(m/z):132(M+).
β-萘氧基丙炔(2c)[22]:白色固体(m.p.=63℃);1H NMR(400MHz,CDCl3)7.78-7.74(m,3H),7.45(t,J=7.2 Hz,1H),7.35(t,J=7.2Hz,1H),7.23(d,J=2.9 Hz,1H),7.20-7.19(dd,J=8.8,2.5 Hz,1H),4.80(d,J=2.4Hz,2H),2.55(t,J=2.4Hz,1H);13C NMR (400 MHz,CDCl3)155.42,134.24,129.56,129.28,127.64,126.90,126.45,123.99,118.70,107.39,78.45,75.63,55.80;Ms(m/z):182(M+).
苯乙 炔(2d)[23]:Oil;1H NMR (400 MHz,CDCl3)7.45-7.41(m,2H),7.26-7.21(m,3H),3.04(s,1H);13C NMR(400MHz,CDCl3)132.22,128.74,128.29,122.12,83.75,77.28;Ms(m/z):102(M+).
对甲苯乙炔(2e)[24]:Oil;1H NMR(400MHz,CDCl3)7.35(d,J=8.2 Hz,2H),7.08(d,J=8.2Hz,2H),3.02(s,1H),2.33(s,3H);13C NMR (400 MHz,CDCl3)138.75,131.83,129.15,119.19,83.77,76.48,21.35;Ms(m/z):116(M+).
1-辛 炔(2f)[25]:Oil;1H NMR (400MHz,CDCl3)2.17-2.19(m,2H),1.96(t,J=2.4 Hz,1H),1.34-1.56(m,12H),0.87(t,J=6.4Hz,3H);13C NMR(400 MHz,CDCl3)84.58,67.95,31.52,29.18,29.15,28.47,22.56,18.34,14.35;Ms(m/z):109(M+).
O,O-二乙基-Se-苯基磷酸酯(3a)[26]:Oil;31P NMR(400 MHz,CDCl3)1H NMR (400 MHz,CDCl3)7.66-7.63(m,2H),7.35-7.31(m,3H),4.23-4.15(m,4H),1.33-1.29(t,J7.00Hz,6H);13C NMR(400MHz,CDCl3)135.2,129.4,128.3,123.7,68.9,15.9;Ms(m/z):294(M+).
O,O-二乙基-Se-对甲苯基磷酸酯(3b)[26]:Oil;
31P NMR(400MHz,CDCl3)1H NMR(400MHz,CDCl3)7.52-7.48(dd,J=8.23Hz,J=1.90 Hz,2H),7.16-7.12(d,J=8.27Hz,2H),4.25-4.19(m,4H),2.36(s,3H),1.34-1.30(t,J7.12 Hz,6H);13C NMR (400 MHz,CDCl3)135.4,129.8,128.4,123.8,68.5,21.5,15.1;Ms(m/z):308(M+).
O,O-二乙基-Se-对氯苯基磷酸酯(3c)[26]:Oil;31P NMR(400MHz,CDCl3)1H NMR(400MHz,CDCl3)7.57-7.54(dd,J8.57Hz,J1.81Hz,2H),7.30-7.27(d,J6.45Hz,2H),4.24-4.17(m,4H),1.36-1.32(t,J7.03 Hz,6H);13C NMR(400 MHz,CDCl3)136.7,130.1,129.4,124.6,67.4,15.5;Ms(m/z):328(M+).
O,O-二乙基-Se-对甲氧苯基磷酸酯(3d[27]:Oil;31P NMR (400MHz,CDCl3)1H NMR(400MHz,CDCl3)7.40-7.36(dd,J=8.20 Hz,J=1.88 Hz,2H),7.05-7.01(d,J=8.27Hz,2H),4.31-4.25(m,4H),3.76(s,3H),1.31-1.27(t,J7.16Hz,6H);13C NMR(400 MHz,CDCl3)155.3,133.9,128.4,119.4,67.8,55.7,15.1;Ms(m/z):324(M+).
2 结果与讨论
以3-苯氧丙炔基苯硒醚与亚磷酸二乙酯的反应为模型(Scheme 3).在室温及氮气下,将1.0 mmol亚磷酸二乙酯与0.2 mmol氢氧化铯加入到5.0mL DMF中搅拌0.5h,然后加入1.0mmol的3-苯氧丙炔基苯基硒醚继续搅拌,TLC 追踪反应进程,发现两个新点,分离并对反产物进行表征,产物为3-苯氧丙炔和O,O-二乙基-Se-苯基磷酸酯.这一结果表明,氢氧化铯催化下,亚磷酸二乙酯并不能对炔硒醚进行加成,而是发生炔硒醚的去硒化反应.

采用上述投料比,空气氛围中,室温下,以3-苯氧丙炔基苯基硒醚与亚磷酸二乙酯的反应为模型,考察溶剂与时间对反应的影响.结果见表1.

表1 溶剂和时间对3-苯氧丙炔基苯基硒醚与亚磷酸二乙酯反应的影响Tab.1 The influence of solvents and time on the reaction for O,O-diethyl phosphonate with alkynyl selenides
从表1知,用二氯甲烷、甲苯作溶剂,在室温几乎不反应,延长反应时间,产率有所提高,但不显著.以乙醇或THF 作溶剂,在室温反应效果比二氯甲烷、甲苯好,但不及DMF与DMSO.由于DMSO 毒性大,所以用DMF作溶剂是一种较佳选择.
氢氧化铯在DMF 与DMSO 中活性大,是因为DMF与DMSO 是偶极非质子溶剂,偶极的负端对铯离子有强的静电力,导致与之键合的阴离子(EtO)2P-(O)之间距离增大,使(EtO)2P-(O)表现出强的亲核性.
随后以DMF做溶剂,采用上述投料比,空气氛围中,室温下反应6h,以3-苯氧丙炔基苯基硒醚与亚磷酸二乙酯为模型,考察催化剂用量对反应的影响.结果见表2.
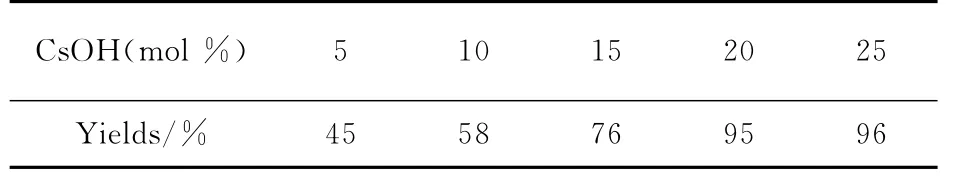
表2 催化剂用量对3-苯氧丙炔基苯基硒醚与亚磷酸二乙酯反应的影响Tab.2 The influence of catalyst amount on the reaction for O,O-diethyl phosphonate with alkynyl selenides
表2表明CsOH 用量为20mol%是较佳选择.
以3-苯氧丙炔基苯基硒醚与亚磷酸二乙酯反应为模型,空气氛围中,室温下,碱的用量为20 mol%,DMF为溶剂,反应6h,探讨不同碱对反应的影响,结果见表3.

表3 不同碱催化下对反应产率的影响Tab.3 Influence of various alkalis on the reaction yields
由表3可知,在相同条件下,CsOH 表现出最好的催化效果.
CsOH 的碱性比表3 中其他碱金属氢氧化物强,是因为铯离子体积最大,与阴离子OH-之间静电力小,使OH-表现出更大活性;同样与Cs+键合的磷负离子(EtO)2P-(O)也表现出强的亲核性.
在上述实验基础上,系统考察不同的炔硒醚与亚磷酸二乙酯的反应(Scheme 4),结果见表4.


表4 端炔和O,O-二烷基-Se-芳基磷酸酯的产率Tab.4 Yields of terminal alkynes and Se-aryl phosphoroselenoates
表4表明,以DMF作溶剂,在20mol% CsOH存在下,不同的炔硒醚与亚磷酸二乙酯均能反应,炔硒醚中芳基电负性上取代基对收率影响不大.
氢氧化铯催化炔硒醚与亚磷酸二乙酯反应可能机理表示如下(Scheme 5):

在上述催化循环中,氢氧化铯与亚磷酸二乙酯反应生成的(EtO)2P-(O)Cs+亲核进攻炔硒醚中的Se生成磷酸硒酯和RC≡C-Cs+,RC≡C-Cs+随后水解得到RC≡CH,同时形成催化剂氢氧化铯.
以无水DMF和含水量0.5%的DMF 作溶剂,结果表明,在含水溶剂中,反应速度较快.
3 结 论
氢氧化铯催化下,亚磷酸二乙酯并不能对炔硒醚进行加成,而是亲核进攻炔硒醚中的硒,生成端炔和磷酸硒酯.本研究的这种意外发现,为硒醚的脱保护提供了一条简便有效的新途径.与文献报道的炔硒醚脱保护方法相比,本方法不使用活泼、不便于操作的试剂,不需要使用易爆的过氧化物,溶剂也无需进行除水处理,仅使用催化剂氢氧化铯,且具有反应条件温和,收率高等优点.
[1]BRAGA A L,COMASSETO J V,PETRAGNANI N.An intramolecular Wittig reaction leading to protected terminal acetylenes[J].Synthesis,1984,3:240-243.
[2]COMASSETO J V,SILVEIRA C C,FERREIRA J T B,et al.The facile deselenation of acetylenic selenides[J].Synthetic Communications,1986,16(3):283-290.
[3]李言杰,曾纪朝,许新华,等.Cp2TiCl2/i-BuMgBr还原断裂Se-Csp键反应研究[J].有机化学,2005,25(10):1227-1229.
LI Yan-jie,ZENG Ji-chao,XU Xin-hua,etal.Cleavage of Se-Csp bond by Cp2TiCl2/i-BuMgBr[J].Youji Huaxue,2005,25(10):1227-1229.(In Chinese)
[4]YOSHIMATSU M,OTANI T,MATSUDA S,etal.Scandium-catalyzed carbon-carbon bond-forming reactions of 3-sulfa-nyl-and 3-selanylpropargyl alcohols[J].Organic Letters,2008,10(19):4251-4254.
[5]SHENG S,LIU X.One-pot synthesis of selenoesters from alkynyl aryl selenides[J].Organic Preparations and Procedures International,2002,34(5):499-502.
[6]TIECCO M,TESTAFERRI L,TEMPERINI A,etal.Synthesis of substituted Se-phenyl selenocarboxylates from terminal alkynes[J].European Journal of Organic Chemistry,2004,16:3447-3458.
[7]PÉREZ-BALADO C,LUCACCIONI F,MARKÓI E.Stereoselective synthesis of(E)-1-iodo-1-selenoalkenes via hydroalumination-iodination of 1-alkynyl selenides[J].Tetrahedron Letters,2005,46(29):4883-4886.
[8]CAI M Z,JIANG M H,LI H G.A facile stereoselective synthesis of(E)-1-arylseleno-2-aryl-sulfanylethenes via hydrozirconation of arylselenoethynes[J].Journal of Chemical Research,2006,11:702-704.
[9]PÉREZ-BALADO C,MARKÓI E.1-Iodo-1-seleno-alkenes as versatile alkene 1,1-dianion equivalents.Novel connective approach towards the tetrahydropyran subunit of polycavernoside A[J].Tetrahedron,2006,62(10):2331-2349.
[10]MANARIN F,ROEHRS J A,GAY R M,etal.Electrophilic cyclization of 2-chalcogenealkynylanisoles:versatile access to 2-chalcogen-benzo[b]furans[J].Journal of Organic Chemistry,2009,74(5):2153-2162.
[11]MANARIN F,ROEHRS J A,BRANDÃOR,etal.Synthesis of 3-alkynyl-2-(methylsulfanyl)benzo[b]furans via Sonogashira cross-coupling of 3-iodo-2-(methylsulfanyl)benzo[b]furans with terminal alkynes[J].Synthesis,2009,23:4001-4009.
[12]YOSHIMATSU M,WATANABE H,KOKETSU E.New cyclization of 4-oxahepta-1,6-diynes bearing sulfur and selenium functional groups[J].Organic Letters,2010,12(18):4192-4194.
[13]LARA R G,BORGES E L,LENARDĀO E J.Addition of thiols to phenylselenoalkynes using KF/alumina under solventfree conditions[J].Journal of the Brazilian Chemical Society,2010,21(11):2125-2129.
[14]OHTA K,OKETSU E K,AGASE Y N,etal.Lewis acidcatalyzed propargylic etherification and sulfanylation from alcohols in MeNO2-H2O[J].Chemical &Pharmaceutical Bulletin,2011,59(9):1133-1140.
[15]AVERSA M C,BARATTUCCI A,BONACCORSI P.Regioand stereocontrolled synthesis of(Z)-α-(Phenylseleno)sulfinyl and-sulfonyl alkenes via sulfenic acids,and a study of their reactivity[J].European Journal of Organic Chemistry,2011(28):5668-5673.
[16]PERIN G,BORGES E L,ALVES D.Highly stereoselective method to prepare bis-phenylchalcogen alkenes via addition of chalcogenolate to phenylseleno alkynes[J].Tetrahedron Letters,2012,53(16):2066-2069.
[17]TAKAHASHI N,NAGASE Y,TANABE G,etal.Synthesis of 3-methyl-and 3,4-dimethylfurans using alkoxide,thiolate,and phenoxide-mediated cyclization of 4-oxahepta-1,6-diynes bearing sulfur and selenium functional groups[J].Tetrahedron,2012,68(5):1566-1580.
[18]夏湘,邹康兵,许新华,等.氢氧化铯催化二硫醚、二碲醚与端炔反应研究[J].化学学报,2008,66(14):1749-1752.
XIA Xiang,ZOU Kang-bing,XU Xin-hua,etal.Study of cesium hydroxide-catalyzed reactions of diaryl disulfides and ditellurides with terminal acetylenes[J].Acta Chimica Sinicn,2008,66(14):1749-1752.(In Chinese)
[19]王小勇,李治章,许新华,等.氢氧化铯催化端炔氢硒化:高立体区域选择性合成(E)-1-芳硒基烯烃[J].有机化学,2013,33(3):558-561.
WANG Xiao-yong,LI Zhi-zhang, XU Xin-hua,etal.Hydroselenation of terminal alkynes catalyzed by cesium hydroxide:highly stereo-and regio-selective synthesis of(E)-1-arylselenoalkenes[J].Chinese Journal of Organic Chemistry,2013,33(3)558-561.(In Chinese)
[20]LI G,ZHAO G.Efficient acetylation of alcohols and phenols catalyzed by recyclable lithium bis(perfluoroalkylsulfonyl)imide[J].Synthetic Communications,2013,43(1):34-43.
[21]QIU W W,SURENDRA K,YIN L,etal.Selective formation of six-membered oxa-and carbocycles by the In(III)-activated ring closure of acetylenic substrates[J].Organic Letters,2011,13(21):5893-5895.
[22]FENG Y S,XIE C Q,QIAO W L,etal.Palladium-catalyzed trifluoroethylation of terminal alkynes with 1,1,1-trifluoro-2-iodoethane[J].Organic Letters,2013,15,936-939.
[23]FUJII A,MILLER S I.Nucleophilic substitution at acetylenic carbon.Kinetics and mechanism of the Arbuzov reaction of substituted phenylbromo-and phenylchloroacetylenes with triethyl phosphite[J].Journal of the American Chemical Society,1971,93(15):3694-3700.
[24]LAMBERT J B,LARSON E G,BOSCH R J.Stereomutation in the Seyferth reaction[J].Journal of the American Chemical Society,1985,107(19):5443-5447.
[25]NELSON D J,BLUE C D,BROWN H C.Hydroboration kinetics.5.Kinetics of the reaction of 9-borabicyclo[3.3.1]nonane with representative haloalkynes in carbon tetrachloride.The effect of halogen substitution upon the stoichiometry and rate of hydroboration[J].Journal of the American Chemical Society,1982,104(18):4913-4917.
[26]XU Q,LIANG C G,HUANG X.Free radical reaction of dialkyl phosphites and organic dichalcogenides:A new facile and convenient preparation of arylselenophosphates[J].Synthetic Communications,2003,33(16):2777-2785.
[27]GAO Y X,TANG G,ZHAO Y F.A novel and general method for the formation of S-aryl,Se-aryl,and Te-aryl phosphorochalcogenoates[J].Synthesis,2009,7:1081-1086.
猜你喜欢
无机盐工业(2020年6期)2020-06-12 03:35:16
山东化工(2020年4期)2020-03-30 08:39:38
石油学报(石油加工)(2019年5期)2019-10-19 07:58:48
世界农药(2019年3期)2019-09-10 07:04:12
安徽化工(2016年4期)2016-11-29 03:40:06
中国粮油学报(2016年1期)2016-02-06 02:16:51
中国资源综合利用(2016年7期)2016-02-03 03:00:19
中国塑料(2015年8期)2015-10-14 01:10:44
营销界(2015年23期)2015-02-28 22:05:26
河北大学学报(自然科学版)(2015年1期)2015-02-27 13:05:59
