
常规基因检测阴性的地中海贫血疑似病例再行进一步基因检测仍有7%的阳性发现
2014-11-21 04:50吕荣钰文飞球陈小文陈仕平
中国循证儿科杂志 2014年4期
吕荣钰 文飞球 陈小文 张 民 陈仕平 李 剑
Hb病包括地中海贫血(简称地贫)和异常Hb病。地贫是由一种或多种珠蛋白肽链合成部分或完全抑制,导致Hb含量异常,以α地贫和β地贫最常见[1]。全球229个国家中,71%的国家受Hb病这一重大健康问题困扰[2]。全球近5亿人为Hb异常基因携带者,每年30~50万新出生儿童为重型的纯合状态;5岁以下死亡儿童中约3.4%死于Hb病[2]。目前治疗Hb病的方法有限,临床实践证实通过早期筛查及遗传咨询来预防和减少Hb病患儿出生是最有效的解决方案,因此明确Hb病的致病基因相当重要。
地贫数据库(http://globin.bx.psu.edu/)资料统计,迄今已发现1 600余种Hb基因异常类型,近10年新发现180余种。临床上根据血液学分析行地贫初筛,确诊仍依赖于基因检测[3]。但在临床工作中发现,部分患儿虽有地贫样表现,血液学初筛阳性,而常规基因检测却呈阴性,不能解释病因,同时临床常规基因检测也可能有一定的漏检率。为此,本研究针对这部分群体再进一步行地贫常规基因和罕见基因的测序和突变类型分析,能进一步明确多少比例的地贫病因,丰富地贫病因学认识,为临床诊断提供参考。
1 方法
1.1 伦理 本研究获得深圳市儿童医院(我院)生物伦理委员会审核同意。
1.2 病例纳入标准 我院地贫血液学初筛阳性且常规基因检测阴性的疑似地贫的连续病例,且获得患者及其家长的书面知情同意再行进一步地贫基因检测。
1.3 地贫血液学初筛 应用CAPILLAPYS2全自动电泳仪(西比亚,法国)进行Hb电泳分析,全自动血液分析仪XS-1000i(希森美康,日本)进行血液分析,UV-5200紫外可见分光光度计(上海元析仪器有限公司)测定葡萄糖-6-磷酸脱氢酶(G6PD)比值及红细胞渗透脆性。①贫血:Hb<该年龄段正常值下限和(或)RBC平均容积(MCV)<80 fl和(或)RBC平均Hb量(MCH)<28 pg;②地贫筛查指标:HbA2和(或)胎儿Hb(HbF)>该年龄段上限值和(或)RBC渗透脆性<0.6;①或②项指标阳性定义为地贫血液学初筛阳性。
1.4 地贫常规基因检测 对于地贫血液学初筛阳性患者,采集外周静脉血3mL(EDTA-K3抗凝),使用基因组DNA快速提取试剂盒提取全血DNA。采用跨越断裂点PCR(Gap-PCR)技术测定3种缺失型 α地贫(-α3.7、-α4.2和--SEA),PCR 反向探针点杂交(reversedot blot,RDB)技术检测3种非缺失型α地贫(Hb Constant Spring、Hb Quong Sze、Hb Westmead),以及17种 β地贫点突变[CD41-42(-TCTT),IVS-2-654(C >T),CD17(A >T),-28(A >G),CD26(G >A),CD71-72(+A),CD43(G >T),-29(A>G),起始密码子 ATG >AGG,CD14-15(+G),CD27-28(+C),-32(C>A),-30(T>C),IVS-1-1(G >T),IVS-1-5(G >C),CD31(-C),CAP+40-+43(-AAAC)]。基因组DNA提取试剂盒以及诊断试剂盒购自深圳益生堂生物公司,由我院儿科研究所专业人员严格按照说明书操作。
1.5 地贫进一步基因检测 地贫血液学初筛指标阳性且我院常规基因检测阴性样本,送深圳华大基因研究所检测。检测除涵盖了上述的常规基因以外,还包括地贫的罕见基因。
对于地贫突变类型,设计3对特异性引物用于检测HBA1、HBA2和HBB 3个基因的突变。每个反应体系为25μL,包含 5μL 基因组 DNA、12.5μL 2 × Gold StarTaq Master Mix(康为世纪)、0.4μmol·L-1引物,在 ABI 9700 PCR 仪(Perkin-Elmer Applied Biosystems,Inc.,Foster City,CA)上完成扩增。实验条件为95℃ 10 min;95℃ 30 s,60℃30 s,72℃ 90 s,35个循环;72℃ 5 min。扩增产物经纯化后在ABI3730xl DNA Analyzer测序仪(Applied BioSystems)上进行正反双向测序,结果采用Chromas和DNAstar软件分析。
对于地贫缺失类型,采用文献报道的Gap-PCR方法检测 α地贫5种缺失型(--SEA、-α3.7、-α4.2、--FIL、--THAI)[4]。同时采用文献报道的荧光定量PCR的方法检测非常见的α和β地贫缺失类型和拷贝数异常[5,6]。
1.6 资料截取 对地贫血液学初筛指标阳性且常规基因检测阴性病例,再进一步行基因检测阳性者,从病历中截取基因检测结果、年龄和血液学筛查结果行描述性分析。
2 结果
2.1 一般情况 2011年1~9月我院地贫血液学初筛阳性878例,其中常规基因检测阴性患者285例(32.5%),年龄10d至34岁;其中0~6月龄18例,~6岁209例,~16岁52例,>16岁6例,平均年龄为(26.5±33.9)个月。
2.2 进一步基因检测结果 21/285例(7.4%)检测阳性,其中常规基因检测阳性5例(1.8%),罕见基因检测阳性16例(5.6%)。21例患者的基因检测结果和临床资料如表1所示。
2.2.1 常规基因突变测序 检出非缺失型β地贫27/28(+C)和41/42(+C)各2例,IVS-II-654(C>T)1例。
2.2.2 罕见基因突变测序结果 ①异常Hb病:1例DNA测序证实在CD91处275T>C(CTT>CCT,Leu>Pro),且为杂合子(图1),查询地贫数据库为异常Hb Port Phillip型 。该例为新生儿,10日龄,血液分析提示小细胞低色素性贫血。检出异常Hb J-Bangkok和Hb Ernz各1例,查询地贫
数据库,Hb J-Bangkok类型为CD56处170G>A(GGC>GAC,Gly>Asp),血常规:RBC 3.94 ×1012·L-1、Hb 113 g·L-1、MCV 83.3 fl、MCH 28.7 pg,均在正常范围。Hb Ernz为CD123处371C>A(ACC>AAC,Thr>Asn)(图2)[7],在亚洲人群中首次发现。本例Hb Ernz携带者为男婴,9月龄,基因测序提示为杂合子,血液学检查结果示:RBC 4.68 ×1012·L-1,Hb 85 g·L-1,MCV 降低(62.6 fl),MCH降低(18.2 pg),HbF正常(0.36%),HbA2 降低(2.36%),铁蛋白降低(4.3 ng·μL-1)。②β基因非缺失型:检出非缺失型β地贫-90(C>T)2例,为罕见β地贫类型。
2.2.3 Gap-PCR和荧光定量PCR分析 ①α基因缺失型:检出非常见缺失型α地贫5例,其中-α/αα 2例、--/αα 3例。血液学检查均呈小细胞低色素性贫血,且为轻度贫血,仅1例HbA2降低,其余Hb电泳正常。②β基因缺失型:检出缺失型β地贫(-/β)3例。血液学检查示小细胞低色素性RBC,无贫血,Hb电泳示HbF明显升高。③α地贫合并β地贫:1例成年女性(例21)检出地贫基因型为--/αα合并-/β,其中α缺失为非常见类型。④α-三联体:2例为α基因三联体(ααα/αα),未合并β地贫,未进一步分型,2例均无贫血(Hb 121~134 g·L-1),RBC呈小细胞低色素状态(MCV 75.5~77.1 fl、MCH 26.2~27.2 pg),Hb电泳未见异常。
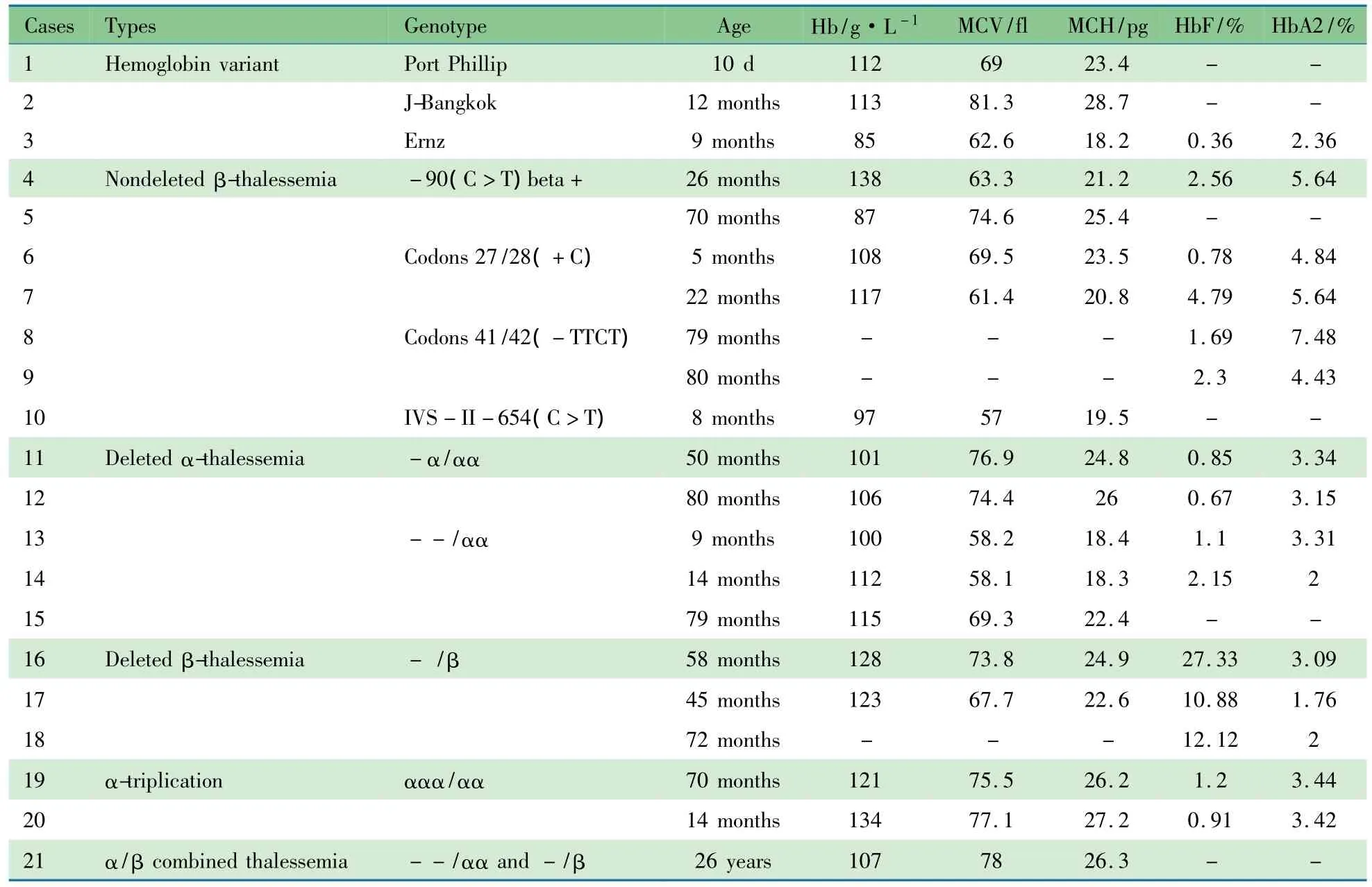
表1 地贫疑似病例再进一步行基因检测阳性21例的基因型和血液学检查结果Tab 1 Genotypes and blood test results of 21 suspected thalassemia cases with negative result in regular genedetection

图1 Hb Port Phillip杂合型突变Fig 1 Hb Port Phillip mutation

图2 Hb Ernz杂合型突变Fig 2 Hb Ernz mutation
2.3 进一步基因检测阴性患者的血液学检查结果 99/264例进一步基因检测阴性患者行血液学检查,66.7%(60/90)铁蛋白低于正常值,其中44例Hb和(或)MCV和(或)MCH低于正常值下限;另有3例提示贫血,但铁蛋白值显著升高(571~1 557 ng·μL-1);8例提示G6PD降低(0.6~0.8)。
3 讨论
本研究结果显示,285例地贫血液学初筛阳性且常规基因检测阴性的患者中,检出21例(7.4%)基因异常,其中常规基因漏诊5例(1.8%),提示目前的常规基因检测方法可能低估了地贫及异常Hb病携带者的数据。Sanger基因测序检出常见β珠蛋白点突变[CD27-28(+C)、CD41-42(-TCTT)、IVS-2-654(C>T)]5例,检出罕见β地贫点突变[-90(C>T)beta+)]2例;荧光定量PCR检出非常见缺失型 α地贫(-α/αα、--/αα)5例,为除-α3.7、-α4.2、--SEA、--THAI和 --FIL以外的罕见缺失类型(如 -α3.5、--CL、--CI等),需进一步分子鉴定以明确分型[7]。因此,对于临床发现和血液学初筛疑似地贫的患儿,即使常规基因检测无异常,仍不能除外Hb病,必要时检测罕见突变类型明确诊断。
本研究首次在亚洲人群发现异常Hb Ernz类型突变(CD123Thr>Asn),丰富了亚洲及中国Hb异常基因种类。文献报道该突变仅在意大利人中检出,位于Hb四聚体外部β链上,对Hb稳定性影响小,可导致RBC增多,机制尚不清楚,需进一步研究[8]。本文检出的该例患儿RBC正常,Hb、MCV、MCH及铁蛋白均降低,可能为婴儿期合并营养性缺铁性贫血所致。
本文1例Sanger基因测序为异常Hb Port Phillip(CD91处Leu>Pro),血液学检查提示为小细胞低色素性贫血。该突变仅见于1977年新西兰1例中国裔女性[9],该突变位于α链,可导致Hb稳定性降低[9]。查询人类野生型Hb X射线结构(PDB 代码:1HHO)[10],采用 Swiss-PdbViewer软件进行三维结构分析得知,α肽链CD91处氨基酸靠近亚铁血红素原卟啉部位,而铁与原卟啉配位结合,且铁可与氧结合;氨基酸的改变可能引起Hb构象改变,影响铁与氧结合,导致稳定性降低和发生溶血性贫血。文献报道中国人群Hb J-Bangkok较常见,且对个体临床表型影响小[11],本文1例测序为Hb J-Bangkok类型。
本研究还检测出2例 α-三联体(ααα/αα),除 RBC 呈小细胞低色素状态,其他筛查指标均正常,提示仅存在此种类型拷贝数增加情况可能对患者Hb功能影响较小。但也有文献报道拷贝数增加可能加剧β地贫患者α链和β链的不平衡趋势,使临床表现更显著[12,13]。
基因测序和PCR结果阴性的264例患者,44/60例铁蛋白降低,且Hb和(或)MCV和(或)MCH降低,考虑营养性缺铁性贫血可能。3例虽有贫血,但铁蛋白值显著增加,可能与炎症等疾病有关,需进一步检测明确病因。8例G6PD比值低于正常值,考虑G6PD缺乏症可能。
本研究提示,对于常规基因检测阴性的疑似地贫患者,仍有1.8%的常规基因漏检率和5.6%的罕见基因突变,因此对这部分人群有必要进一步行常规基因的复检和罕见基因的检测。
本研究不足和局限性,检出的地贫非常规基因突变的病例数较少,尚不能建立基因型和表型之间的关系,待扩大样本后进行分析,对于临床表型有针对性的进行相应基因的检测。
[1]胡亚美,江载芳.主编.诸福棠实用儿科学.第7版.北京:人民卫生出版社,2002,1763-1774
[2]Thalassemia International Federation.Guidelines for the clinical management of thalassemia.2nd edition.2008:21-27
[3]Zhou YQ(周玉球).Current situation and prospect of hematological phenotypic screening and genotyping of thalagsemia.Chinese Journal of Laboratory Medicine(中华检验医学杂志),2012,35:394-398
[4]Chong SS, Boehm CD, Cutting GR, et al.Simplified multiplex-PCRdiagnosis of common southeast asiandeletional determinants of alpha-thalassemia.Clin Chem,2000,46(10):1692-1695
[5]Fallah MS, Mahdian R, Aleyasin SA, et al.Development of a quantitative real-time PCR assay fordetection of unknown alpha-globin genedeletions.Blood Cells Mol Dis, 2010,45(1):58-64
[6]Babashah S, Jamali S, Mahdian R, et al.Detection of unknown deletions in beta-globin gene cluster using relative quantitative PCR methods.Eur J Haematol, 2009,83(3):261-269
[7]曾溢滔.主编.人类血红蛋白.第1版.北京:科学出版社,2002,85
[8]Groff P,Kalmes G, Golinska B, et al.Hb Ernz[beta123(H1)Thr> Asn] and Hb Renet[beta133(H1)Val>Ala]:Two new neutral vatiants revealed by reversed phasehigh performance liquid chromatography analysis.Hemoglobin,2000,24(4):287-297
[9]Brennan SO, Tauro GP, Melrose W. Haemoglobin Port Phillip alpha91( FG3) Leu replaced by Pro,a new unstable haemoglobin. FEBS Lett, 1977, 81( 1) : 115-117
[10]Shaanan B.Structure of human oxyhaemoglobin at 2.1 A resolution.J Mol Biol, 1983, 171(1):31-59
[11]Zhao Y(赵颖), Shang X, Xiong F, et al.Analysis of clinical phenotypes of compound heterozygotes of Hb J-Bangkok and βthalassemia.Chinese Journal of Medical Genetics(中华医学遗传学杂志),2013,30(2):148-151
[12]Giordano PC, Bakker-Verwij M, Harteveld CL.Frequency of a-globin gene triplications and their interaction with bthalassemia mutations.Hemoglobin,2009,33(2):124-131
[13]Panigrahi I, Mahapatra M, Kumar R, et al.Jaundice and alpha gene triplication in beta-thalassemia:association or causation?.Hematology, 2006, 11(2):109-112
猜你喜欢
中国毕业后医学教育(2022年1期)2022-08-19
昆明医科大学学报(2021年5期)2021-07-22
中华养生保健(2020年3期)2020-11-16
食品与健康(2020年3期)2020-03-28
中国乳品工业(2019年10期)2019-12-09
World Journal of Clinical Cases(2019年6期)2019-04-17
农产品加工(2019年8期)2019-01-06
读与写·下旬刊(2018年12期)2018-12-19
家庭用药(2016年7期)2016-05-14
江苏农业科学(2016年3期)2016-05-03
