
快速催化极谱法测定土壤中的有效态钼
2014-11-20 05:14陈志慧孙洛新钟莅湘尚保忠
岩矿测试 2014年4期
陈志慧,孙洛新,钟莅湘,王 琰,尚保忠,肖 抒
(1.河南省地质调查院,河南郑州450001;2.河南省岩石矿物测试中心,河南郑州450012)
在国土资源地质大调查中,生态地球化学评价得到了足够重视,大批量样品的测试对实验室带来了巨大挑战和压力。土壤中元素有效态是生态地球化学评价的重要内容之一,其分析大多按照相应的国家、行业或地方标准进行[1]。但在实际工作中发现有些方法测试效率低,结果波动大,难以进行大批量样品的分析,因此研究开发快捷、经济有效的分析测试方法显得尤为重要。
以钼的分析为例。钼是植物生长必需的微量元素之一,我国土壤中全钼的含量为0.1~6 mg/kg,平均含量1.7 mg/kg,有效态钼的含量范围为0.02~0.5 mg/kg[以草酸-草酸铵(Tamm溶液)为浸提剂]。土壤中的钼可分为四部分:水溶性钼(含量少);交换性钼(被土壤矿物颗粒或铁锰的氧化物吸附的MoO2-4,对植物有效);矿物态难溶性钼(对植物无效);有机结合态钼(与土壤有机质结合的钼,随着有机质矿化,可以被释放出来为植物所吸收利用)。土壤中有效态钼主要指水溶性和交换性钼[2-3],其测定方法有多种化学法和仪器方法。比色法[3-6]灵敏度较高,但显色条件要求严格,分离富集手续繁琐;火焰原子吸收光谱法测定钼时,仅有部分钼被原子化,测定的灵敏度较低;近年来应用石墨炉原子吸收光谱法[7-8]、电感耦合等离子体发射光谱法[9]和电感耦合等离子体质谱法可省去冗长的分离富集操作,但是仪器昂贵[10-11],使用费用高,且检出限偏高(0.0x μg/g),不适于在一般实验室普及,与中国地质调查局关于生态地球化学评价样品分析技术要求(有效态钼检出限0.005 μg/g)相差较远[12]。
草酸-草酸铵溶液(Tamm溶液)浸提催化极谱法是测定土壤中有效态钼的经典方法,仪器成本低、干扰少、灵敏度高、检出限低(0.00x μg/g),且测定结果准确稳定。该方法编入国家标准(GB/T 7878—1987)[5]、林业行业标准 LY/T 1259—1999[6]及农业行业标准(NY/T 1121.9—2006)[13]等。目前对土壤中有效态钼的提取也是以草酸-草酸铵溶液(Tamm溶液)的应用最为广泛[3-11,13-20]。我国土壤酸碱性大多在pH 4.5~8.5之间,个别北方地区的pH值高达10.5,南方有的地区pH值低至3.6[3]。该种草酸盐溶液的缓冲容量较大,适用于pH 3.6~10.5的土壤有效态钼的提取。但是浸提液中的铁、锰共存离子影响钼的极谱波形和波高,草酸盐及有机质大量存在严重降低钼极谱波的灵敏度,由于土壤中有效态钼的含量很低,导致测定难以进行。为消除这种干扰,标准方法多采用阳离子交换树脂消除铁、锰等共存离子,干法灰化法(450℃灼烧4 h)消除草酸盐和有机质,操作较为繁琐。本文针对此方法操作过程中的干扰问题,在草酸-草酸铵浸提液中加入固体氢氧化钠沉淀分离铁、锰等杂质,然后以硝酸-硫酸分解破坏浸提液中的有机质及草酸盐,利用钼-苯羟乙酸-氯酸盐-硫酸体系极谱催化波进行测定,实现了土壤中有效态钼的快速测定。
1 实验部分
1.1 仪器及工作条件
JP-2示波极谱仪(成都分析仪器厂),仪器工作条件:参比电极用饱和甘汞电极,原点电位旋钮置于0 V,电极开关用三电极,测量开关用阴极化,电解开关用阳极化,导数开关选用导数。
HY-8A大容量振荡器(金坛市顺华仪器有限公司)。
1.2 标准工作溶液
钼标准储备溶液:称取经500℃灼烧过的氧化钼(MoO3,优级纯)0.1500 g溶解于0.1 mol/L氢氧化钠溶液10 mL中,用去离子水稀释定容至1 L。即为100 μg/mL钼标准储备溶液。
钼标准溶液:吸取100 μg/mL钼标准储备溶液10.00 mL定容至1 L,即为1 μg/mL的钼标准溶液。根据需要逐级稀释至0.1 μg/mL钼标准溶液和0.01 μg/mL钼标准溶液。
1.3 主要试剂
草酸-草酸铵浸提剂的配制:称取24.9 g草酸铵与12.6 g草酸溶于水,定容至1000 mL。酸度为pH=3.3,必要时定容前用pH计校准。
氢氧化钠(分析纯,洛阳昊华化学试剂有限公司),须检验钼空白值。
硝酸(优级纯,ρ=1.42 g/mL,洛阳昊华化学试剂有限公司)。
50%硫酸(分析纯,洛阳昊华化学试剂有限公司)。
100 g/L苯羟乙酸(苦杏仁酸)溶液。
60 g/L氯酸钠溶液。
氯酸钠-苯羟乙酸混合底液的配制:60 g/L氯酸钠溶液1000 mL和100 g/L苯羟乙酸溶液100 mL混合均匀。
1.4 样品分析
1.4.1 样品分析步骤
称取通过1 mm孔径筛的风干土壤试样5.00 g(精确至0.01 g),置于100 mL塑料瓶中,加入50 mL草酸-草酸铵浸提剂,塞严瓶塞后,置于(25±3)℃的环境条件下,振荡1 h后放置过夜。次日移取20 mL清液于50 mL烧杯中,加入1.0 g氢氧化钠,摇匀后放置澄清。吸取清液5 mL,置于25 mL烧杯中,加入1 mL 50%的硫酸、2 mL硝酸,在电热板上低温蒸至硫酸刚刚冒烟,取下冷却至室温,加入10 mL蒸馏水、11 mL混合底液,混合均匀放置半小时后选择合适的电流倍率在极谱仪上测量峰高,其峰值电位为-0.22 V。
1.4.2 空白溶液制备
100 mL浸提液中加入5.0 g固体氢氧化钠,搅拌至氢氧化钠溶解,随样品一起放置澄清,备用。
1.4.3 结果计算
土壤有效态钼含量(mg/kg)的计算公式如下:
式中:m1—标准曲线查得Mo的质量(μg);m—土壤样品质量(g);tS—分取倍数,浸提时所用浸提剂的体积(mL)与测定时吸取浸出液的体积(mL)之比。
2 结果与讨论
2.1 浸提方法的选择
土壤中有效态钼普遍选用草酸-草酸铵溶液作浸提剂,常见浸提方法一是振荡8 h干过滤[5-6],二是振荡6 h后离心取清液[7],三是振荡0.5 h放置过夜后干过滤[13],四是振荡1 h放置过夜,次日吸取清夜[15]。为方便批量样品分析,本文选用第四种浸提方式。(不用振荡器的情况下可先将土壤样品和浸提剂混合均匀,然后间隔0.5~1 h振荡摇匀一次,振荡4~5次后放置过夜,次日不经过滤直接吸取清液也可达到很好的浸提效果)。选择三种方式作了对比,表1结果表明样品和标准物质有效态钼的分析结果无明显差异。
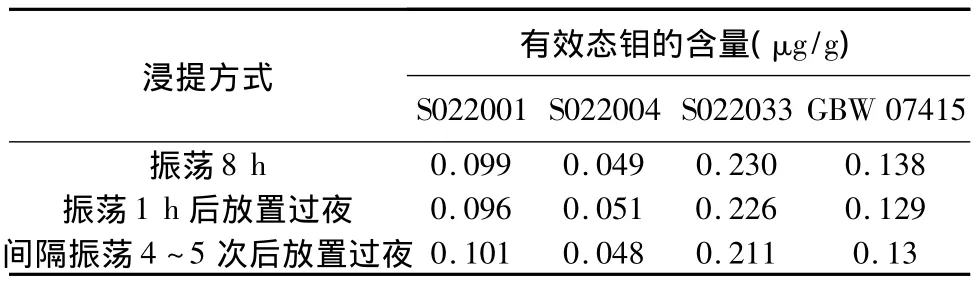
表1 不同浸提方法比较Table 1 Comparison of extraction methods
2.2 浸提温度
温度是影响土壤中钼吸附和解吸的重要因素之一[2],由于用浸提剂浸提土壤有效态钼属于非平衡反应,所有影响浸提剂与土壤间反应速率的因子,如土壤样品的粉碎程度、液土比、浸提温度等,都会影响到土壤有效态钼的浸提效果,因而必须固定提取条件,否则测定结果就无法参与评价。本文严格按照行业标准和国家标准规定的土壤有效态钼浸取温度进行操作,温度控制在25℃,允许有 ±3℃的波动。
2.3 干扰的消除
2.3.1 固体氢氧化钠消除铁和锰共存离子干扰
草酸-草酸盐浸提剂既能浸提土壤中有效态钼,也能浸提土壤中的铝、铁、锰、镁、钾、钛、钒、磷、铜、铅、锌、镓、锡、铌、钽、钨、钇组稀土等离子[16]。试验表明多数共存离子量很小,一般达不到干扰测定的量。铝、镁、钾、磷等元素的离子含量较多,但是这些离子性质稳定,不干扰钼的测定。铁、锰、钛等多价态的离子易发生氧化还原反应,一方面其还原态会消耗体系中与峰电流几乎呈正比关系的氯酸根离子,另一方面这些离子在氧化剂氯酸根离子存在下也会发生类似的平行催化电极反应,影响钼的波形和波高。
文献[16-19]对铁和锰离子干扰钼测定的问题进行了试验研究,尽管使用的极谱仪型号不同,采用的极谱波类型不同,但都表明铁和锰离子达到一定量时会严重影响钼的测定。同时也表明酸性土壤Tamm浸提液中铁和锰等干扰离子的量远远大于它们的允许值,测定前必须予以消除。文献采用的氢氧化锰共沉淀法[16]、732型强酸性阳离子交换树脂法[17]、硝酸 - 高氯酸法[18]和氨水分离法[19]都较好地消除了铁和锰等离子的干扰。石灰性土壤Tamm浸提液中铁、锰等干扰离子的量在允许值以下,可以不予分离[5-6],考虑到样品分析条件的一致性,对中性、酸性及石灰性土壤统一进行干扰离子消除。
本文选用固体氢氧化钠作为沉淀剂分离铁、锰离子。于25 mL浸提液中加入5 mg的Fe3+和1 mg的Mn2+,然后分别加入不同量的固体氢氧化钠,实验现象见表2,取清液酸化后用原子吸收光谱法测量铁和锰残余量。试验表明,采用浸提清液直接加0.5 g固体氢氧化钠,溶液碱度偏低,铁、锰沉淀不够完全;加1.0 g和1.5 g固体氢氧化钠,溶液的pH>13时可完全分离铁、锰等杂质,因此本文氢氧化钠的用量为1.0 g。

表2 固体氢氧化钠用量试验Table 2 Dosage tests of solid natrium hydroxide
2.3.2 硝酸-硫酸混合酸消除草酸盐和有机质干扰
Tamm溶液的还原性来源于草酸,其标准电位为-0.49 V,在酸性溶液中能够还原氯酸根离子,由于浸提液中草酸的大量存在,将大大降低测定液中氯酸根离子的浓度,因此导致测定灵敏度大幅降低,这一现象在戴自强[18]的试验中得到证实。从土壤中浸取出的有机质,是具有复杂结构的酸性高分子化合物,具有明显的还原性;另外有机质多为疏松多孔的胶体,具有强吸附性,其存在会改变溶液的黏度进而影响钼络离子的扩散速度,导致峰电流偏低。传统方法多采用干法破坏,即低温蒸干,然后高温炉450℃灼烧4 h[5-6],或高温炉 500℃灼烧 2 h[9];也有用高氯酸 -硝酸氧化消除法[13,18]、高氯酸 -硝酸 -硫酸氧化消除法[20]。这些方法耗时耗能且结果波动较大。
硫酸冒烟时温度达338℃,而草酸在189.5℃或者遇到浓硫酸就会分解,H2C2O4=CO2+CO+H2O;汤普森等[21]认为硝酸-硫酸是无机元素测定之前破坏有机质最常用的方法。基于此,本文采用加入1 mL 50%硫酸溶液、2 mL硝酸,在电热板上低温蒸至硫酸刚刚冒烟的方法消除草酸盐和有机质干扰。如果分离铁、锰后的溶液为黄褐色,说明有机质含量高,可以适当加大硝酸用量。该方法不需灼烧、蒸干,也不需要加指示剂调酸度等步骤就能很好地控制硫酸的用量,而且有机质分解完全,大大缩短了分析时间,提高了分析速度。
2.4 硫酸的用量及作用
硫酸浓度在0.09~0.36 mol/L[22]时钼波既灵敏又稳定,本文中硫酸有两个作用:一是氧化作用,与硝酸一起消化有机质和草酸盐;二是控制测定溶液的酸度。实验选择硫酸用量为50%硫酸1 mL,蒸至硫酸刚刚冒烟时取下,测定体系中硫酸浓度大约为0.30 mol/L。
2.5 方法技术指标
2.5.1 标准曲线和方法检出限
分别吸取 0.01 μg/mL 钼标准溶液 0.0、0.5、1.0、2.0 mL,0.1 μg/mL 钼标准溶液 0.5、1.0、2.0 mL 和1 μg/mL 钼标准溶液 0.5、1.0、2.0 mL,置于25 mL烧杯中,各补加1.4.2节空白溶液5 mL,配制成0、0.005、0.01、0.02、0.05、0.10、0.20、0.50、1.00、2.00 μg钼标准系列,以下同1.4.1节样品分析步骤。以测得的峰高与对应的钼量(μg)作标准曲线,钼量在0~1.00 μg范围内呈良好的线性关系,相关系数为0.9995。本方法可测定的有效态钼含量范围为0.005~2 mg/kg。
按照本文方法进行12份样品空白溶液的测定,经计算得到测定值的标准偏差(s)为0.000253,考虑到称样量和定容体积,以3倍标准偏差计算得到方法检出限为0.0015 μg/g,满足中国地质调查局《生态地球化学评价样品分析技术要求》对有效态钼的检出限要求(0.005 mg/kg)。而采用林业行业标准方法处理12份样品空白溶液,测得钼的检出限为0.0068 μg/g(按3倍标准偏差计算),高于本法检出限。
本文定量加入固体氢氧化钠沉淀铁、锰离子,简化了操作;硝酸-硫酸混合酸消化草酸盐和有机质的方法,缩短时间不低于5 h;定量加入硫酸,控制体系酸度,免去了加指示剂调节酸度等步骤,在降低消耗简化操作手续的同时降低了空白值、减少了误差来源,使方法检出限明显降低。
2.5.2 方法准确度
选用4个不同酸碱度的国家一级土壤标准物质GBW 07413~GBW 07416,按照本文方法和林业行业标准方法进行准确度试验,测定结果见表3。从分析结果可以看出,本文方法相对误差均小于8%,测定值和标准值更加吻合。
2.5.3 方法精密度
选择3个不同含量范围的土壤样品进行精密度试验,按照本文方法平行分析12次测定有效态钼的含量,计算的精密度(RSD)<7%(表4)。
3 结语
土壤浸提液中的铁、锰等离子、草酸盐和有机质具有还原性,消耗测定体系中与峰电流几乎呈正比关系的氯酸根离子浓度,降低催化波的灵敏度,对测定造成严重干扰,因此必需将浸提液进行测定前处理。本文采用固体氢氧化钠沉淀分离铁、锰等离子,硝酸-硫酸破坏草酸盐及有机质,有效地消除了干扰,克服了传统方法手续冗长等缺点。由于所用试剂种类少,处理流程短,有效地降低了误差来源和空白值,方法检出限由传统方法的0.0068 μg/g降低至0.0015 μg/g,提高了分析结果的精密度和准确度,标准物质有效态钼的测定值和标准值更加吻合。本方法经上千件土壤样品的验证,适用于pH 3.6~10.5土壤中有效态钼的测定,可测定范围为0.005~2 mg/kg,结果稳定可靠,实为一种快捷、经济有效的分析测试方法。
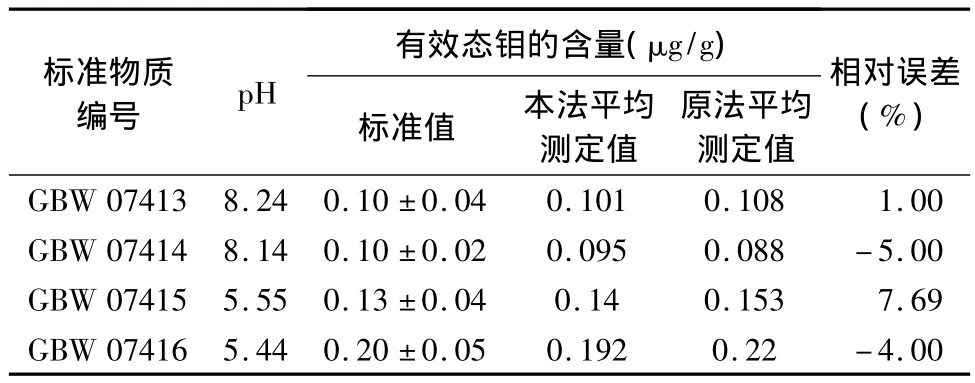
表3 方法准确度Table 3 Accuracy tests of the method
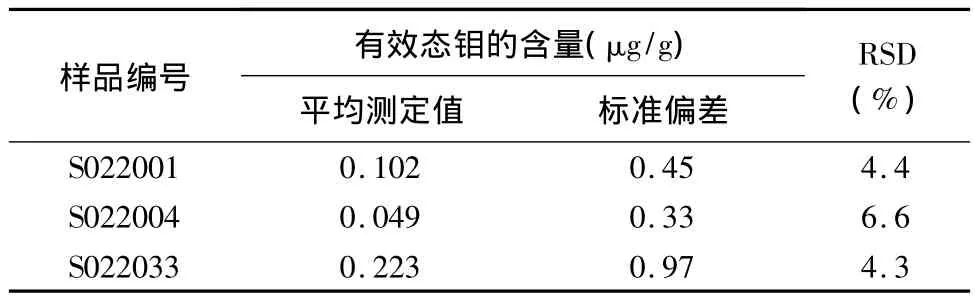
表4 方法精密度Table 4 Precision tests of the method
由于土壤中有效态钼的含量很低,有些厂家氢氧化钠产品中钼的空白值较高,需严格检验所用试剂的空白;极谱催化波受温度影响较大,极谱测定应在同一温度下进行,待测液加入混合底液后温度增高,需放置到室温测定。
[1]周国华.多目标区域地球化学调查:分析测试面临的机遇和挑战[J].岩矿测试,2010,29(3):296-300.
[2]刘鹏,杨玉爱.土壤中的钼及其植物效应的研究进展[J].农业环境保护,2001,20(4):280-282.
[3]鲍士旦.土壤农化分析[M].北京:中国农业出版社,2000:115,141-149.
[4]杨雪兰,樊亚东,苏云松.土壤中有效钼测定方法的研究进展[J].理化检验(化学分册),2010,46(12):1485-1487.
[5]GB/T 7878—1987,森林土壤有效钼的测定[S].
[6]LY/T 1259—1999,森林土壤有效钼的测定[S].
[7]邱海鸥,杨小秋,周延彪,汤志勇.土壤中钼的形态分析方法研究[J].安全与环境工程,2002,9(4):6-7.
[8]苏启英,张健康,袁宇,杨妍虹,苏子奇.石墨炉原子吸收光谱法测定土壤中有效钼[J].理化检验(化学分册),2009,45(3):359,361.
[9]孙朝阳,贺颖婷,王雯妮.端视电感耦合等离子体发射光谱法测定土壤中有效钼[J].岩矿测试,2010,29(3):267-270.
[10]岩石矿物分析编委会.岩石矿物分析(第四版第四分册)[M].北京:地质出版社,2011:891-892.
[11]李力争,吴赫,韩张雄,王龙山,陶秋丽.电感耦合等离子体-质谱法测定土壤中的有效钼[J].光谱实验室,2012,29(4):2282-2285.
[12]中国地质调查局.DD2005-03,生态地球化学评价样品分析技术要求(试行)[S].
[13]NY/T 1121.9—2006,土壤有效钼的测定[S].
[14]李锡坤,李鉴伦,洪翔,冯超,何平.酸性土壤中20种元素有效态浸提体系浅析[J].岩矿测试,2001,20(3):167-172.
[15]钱玥,许洪祥.土壤中有效钼的快速测定[J].黑龙江国土资源,2006(11):42-44.
[16]付爱瑞,肖凡,罗治定,查晓康,孙连伟.氢氧化锰共沉淀分离-催化极谱法测定土壤中有效钼[J].理化检验(化学分册),2012,48(4):417-419.
[17]徐俊样,朱其清.土壤中有效相的极谱(催化波)测定方法[J].土壤学报,1983,20(2):197-204.
[18]戴自强.土壤中有效相的示波极谱测定[J].土壤学报,1986,23(1):82-88.
[19]许庆福,王卿.催化极谱法测定土壤中有效钼方法的改进[J].理化检验(化学分册),2004,40(3):173,175.
[20]万耀星,刘雄德,李正艳.土壤有效钼及植物全钼的示波极谱测定[J].土壤通报,1988,19(1):43-46.
[21][英]汤普森M,沃尔什J N.ICP光谱分析指南[M].北京:冶金工业出版社,1983:246-247.
[22]叶家瑜,江宝林.区域地球化学勘查样品的分析方法[M].北京:地质出版社,2004:372.
猜你喜欢
化工生产与技术(2022年3期)2022-07-01
初中生学习指导·中考版(2021年2期)2021-09-10
肾脏病与透析肾移植杂志(2021年1期)2021-01-13
中小企业管理与科技(2019年18期)2019-08-06
中国医药指南(2018年30期)2018-11-16
东华大学学报(自然科学版)(2016年4期)2017-01-10
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
中学生数理化·中考版(2015年12期)2015-09-10
食品工业科技(2014年13期)2014-03-11
