
低质量数元素同位素在线连续流同位素比值质谱分析的质量控制和数据标准化
2014-11-20 05:13查向平郑永飞
岩矿测试 2014年4期
查向平,龚 冰,郑永飞
(中国科学院壳幔物质与环境重点实验室,中国科学技术大学地球和空间科学学院,安徽合肥230026)
在过去几十年中,稳定同位素分析应用于众多的研究领域,如农业、环境、海洋、临床和司法等,且这种应用成果呈指数增长。20世纪60年代以前,稳定同位素组成的测量和应用很大程度上仅局限于同位素地球化学领域,当时可用的质谱需要量身定做来满足专业化的需求[1],而且这些仪器也仅为一些专职技术人员操作使用,输出的结果也仅被少数的专业学者理解[2]。近些年来,质谱不仅发生了革命性变化,而且出现了多家仪器制造商,如ThermoFisher公司(原 Finnigan 公司)、IsoPrime(原VG公司)、Cerson公司、Nu公司等,使之成为众多同位素实验室可配置、易使用的常规方法。最初这些仪器的设计是为了测量相对小的同位素比值变化(δ值变化小于1‰),而同时具有高的精度和准确度(±0.01‰~±1‰)。尽管那时的质谱装配实现了部分自动化,但在一些情况下样品的测试效率仍然维持在每天约10个样品,很大程度上这是由于复杂的非在线样品制备过程引起的。
20世纪80年代早期,一些研究者为了提高分析效率,加快了简单快速地分析低质量数气体同位素方法的研制步伐。连续流同位素质谱方法是此时发展并很快得以应用的一种较新的革新方法之一。近年来,连续流质谱系统在国外发展很快,先前研究者陆续地介绍了这种系统并为提高这种系统所测数据的可靠性作出了努力。Brenna等[3]就对高精度的连续流同位素质谱作了系统阐述。Midwood等[1〛总体介绍了连续流同位素质谱对碳、氢、氧、氮和硫同位素的分析进展。往后一些研究者又对连续流同位素质谱就提高数据精度和准确度、实现不同实验室之间的数据可比性开展了一些研究[4-16]。目前,我国使用连续流方法对稳定同位素比值的分析和测定报道较多[17-25]。王政等[17]利用元素分析仪-同位素比值质谱(EA-IRMS)对土壤样品中氮同位素进行分析,指出载气、氧气的流量,并就测定中出现的累积效应和氧化能力降低的问题进行了探讨。曹建平等[18]就海洋悬浮颗粒氮同位素进行EAIRMS测定,指出了该方法在一定离子流强度和同位素比值的质谱线性问题。崔杰华等[19]通过多组实验对比,分析并讨论了利用EA-IRMS联用技术测定植物样品中碳同位素比值的实验条件,初步建立了植物样品中稳定碳同位素组成的分析方法,同时对系统分析的稳定性和精密度等进行了检验分析。王旭等[20]给出了EA-IRMS联机系统的燃烧转化率随测量样品次数的变化规律,以及样品的氮、碳同位素测量值与其燃烧转化率的关系等。就目前文献来看,大部分仅对连续流方法中个别方面具体分析,但就如何构建连续流同位素质谱(EA-IRMS、TC/EA-IRMS)系统方法研究尚不多见,尤其是连续流系统中质量控制和数据处理更鲜有报道。
虽然连续流方法可以实现样品制备和气体质谱分析一次性快速地完成,但是一般主要用于复杂和微量以及原位样品的同位素分析,要保证得到可靠的同位素数据(即从准确度、精度、重现性等方面考虑),这不仅需要好的分析策略和运行方案,还需要对仪器日常性能和数据质量进行严密的监视管控,而且还取决于原始数据如何进一步标准化到国际同位素尺度上一套协议。正如Werner等[26]所指出,载气和联线同位素比值分析系统(即连续流系统)覆盖了很大范围,在全世界利用其测量稳定同位素比值的应用越来越广泛。尽管整体上在这个科学领域大有好处,但也引起了一系列的问题。我们感觉到最初少数专家拥有的知识水平经过多年相当程度地冲淡了,结果同位素比值的整体精度和可靠性可能下降了。尤其值得注意的是,报道的δ值相对于国际标准的准确度在分析中产生了大的偏差。因此,保证连续流系统获得高精度和准确的同位素比值,建立与之相关的操作协议方案,质量控制及数据处理步骤就显得尤为迫切。
连续流系统设备的安装和环境控制、测试准备、样品制备和称量、标准物质选择及序列所处外围环境,调节灵敏度和线性,背景值监测,稳定性检测,系数校正等质谱自身属性相关的质量控制,以及所用的标准物质策略、原始数据的标准化以及数据处理等,这些因素对最终的同位素测试结果都有重大影响,本文对这些方面作出总结和评述,有助于同位素质谱的新用户对于该种技术的相关方法和技巧有明确的初步认识和理解。
1 连续流系统
1.1 连续流质谱
“连续流质谱”是相对“双路进样质谱”而言的,它是在气体同位素质谱基础上发展起来的一种新型仪器。先前研究者也称为同位素比值监测质谱(IRM-MS)[26]。连续流同位素比值质谱的萌芽应该说是源于早期的有机质谱,它结合了高效的色谱分离技术和高灵敏的质谱检测技术优势,具有减少样品损失和无可比拟的灵敏度、高效和实用性等优点。连续流质谱方法是非常理想地结合了色谱技术和同位素比值质谱的一种方法。气相色谱及以后的其他分离装置和质谱之间的接口,主要是使用整体结构连续输送分析物气体进入质谱进行分析。无论这些系统多么复杂,都是依赖载气传输分析物,经过在线化学转化阶段,或先通过在线转化分析物为气体,再通过载气输送以适合质谱分析,这个过程就是连续流质谱的基本原理之一[3]。所谓“连续流”是与使用的载气相关的概念,载气把制备系统中的样品气输送到质谱。一般载气为He气,因为He在化学性质上为惰性并且很容易得到高纯度的气体。第一台真正意义上的连续流同位素质谱是Preston和Owens[27]建立起来的,他们将一台专门分析氮元素的分析仪连接到气体同位素比值质谱的离子源上。在Preston和Owens首次成功实现了15N和全氮的高精度和快速分析后不久,又实现了对固体样品转化为CO2后的13C/12C分析。现代的连续流同位素比值质谱仪更加紧凑,自动化程度更高,在几分钟内能够同时分析同一个样品的两个或更多同位素。
1.2 常见的连续流系统
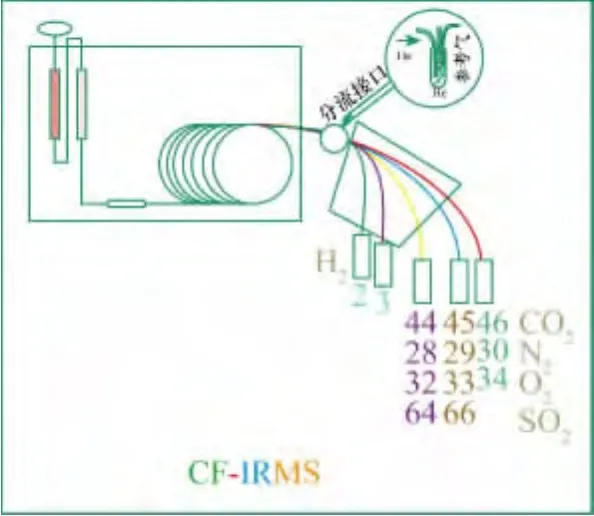
在样品进行同位素比值质谱分析之前,样品首先要无分馏地转化为质谱易接收和方便测量的简单气体,例如,测量2H/1H、13C/12C、15N/14N 和18O/16O这些轻元素同位素的比值,就需要将样品转化为H2、CO2、N2和CO气体等,这取决于物质的组成和所要研究的同位素。同位素比值质谱测量的离子比值与这些气体是分别对应的。目前,轻稳定同位素分析最为常用的连续流装置为元素分析仪与同位素质谱的联用,它可分为两种:一种为EA-IRMS,可用于碳、氮和硫元素同位素分析,样品在氧气中高温燃烧,是一种氧化过程。另一种为高温转化TC/EA-IRMS,主要用于氢和氧的分析,样品主要是在还原环境中经历高温热转化。两种联用系统见图1。
1.2.1 EA-IRMS系统
元素分析仪(EA)一般由两个反应器——燃烧反应器和还原反应器构成,这些反应器可以是两个反应管,也可以结合在单反应管中。反应器之后是水分离装置和填充了分子筛的气相色谱柱(Poropak®QS),色谱柱主要用来分离生成的气体。元素分析仪和质谱之间通过一个分流接口连接。就碳、氮分析而言,其燃烧和气体转移过程如下。
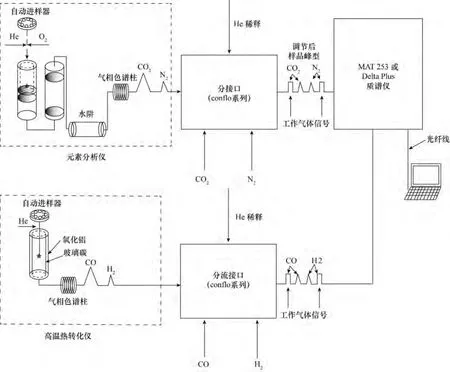
图1 EA-IRMS和TC/EA-IRMS系统简图Fig.1 The diagrams of EA-IRMS and TC/EA-IRMS systems
样品首先在氧气氛围下的反应器中燃烧,生成了CO2、NOx和H2O。尽管对于各种特定应用所推荐的反应管充填材料有很大变化,但反应器一般包含Cr2O3和Co3O4+Ag(主要用来吸附硫和卤素,银还可能起到催化作用)。反应器的温度一般设置在900~1020℃,但是,锡杯的燃烧释放出的燃烧热大约能够提高温度到1800℃(“flash”燃烧)。推荐使用石英嵌入体(灰坩埚或屑坩埚),用来收集灰烬以及可能残留的样品和锡杯,这样在分析了50~150个样品后要进行更换嵌入体(这取决于使用的嵌入体的类型),而此时不必更换反应管。在650℃的第二反应器(还原反应器)中去除过量的O2并将NOx还原为N2。一般填充物为高纯铜,但对于不同的应用所推荐填充料有所变化。水在含有高氯酸镁(无水高氯酸镁)的水阱中分离。如果仅对氮同位素进行同位素比值测定时,使用含有碱石灰、烧碱石棉(硅基质上含有NaOH)或Carbosorb®化学阱将CO2从气体流中去除是最优化的方法。最后,N2和CO2在加热的色谱柱中进行分离(例如,Porapak®QS 50/80,3 m×6.5 mm)。作为色谱柱分离的一种替代方法,一些仪器使用了一个“吹扫捕获器”系统来进行分离。“吹扫捕获器”工作原理为:氮气直接通过系统而其他生成的气体在多个吸附管中收集(通过短的但有分离效果的气相色谱柱实现)。这些吸附管中气体通过电子加热器来释放。
EA-IRMS系统主要用于碳和氮分析[17-23],还可对硫同位素进行分析[6-11],在实际使用中可应用于多种物质。对于固态物质和非挥发性液体使用锡舟(锡杯,用于C、N分析)或银杯(H、O分析)引入到元素分析仪系统,而有限黏度的液体可用液体进样器直接注入到反应器中。
EA-IRMS方法可以实现样品制备和质谱分析一次完成,简化了转化步骤,从而减少了分馏环节。样品的制备简单,不需要或很少需要化学或机械矿物相分离,样品气体的直接引入实现了快速分析,大大地提高了获取稳定同位素数据的能力[28];样品用量少,纯度要求不高,适用样品较广[21]。因此,EA-IRMS系统在医药、食品和农业研究上早已广泛使用,并且其发展正向生态、环境和地球科学领域渗透。
1.2.2 高温热转化TC/EA-IRMS系统
TC/EA-IRMS主要用于氢和氧分析,高温转化温度一般在1350~1450℃。有机和无机化合物转化为H2、N2和CO气体。系统一般由一个由熔融氧化铝制造的外管和一个由玻璃碳组成的内管构成。内管中填充有玻璃碳颗粒和用来吸附卤素的银丝。和EA-IRMS一样,不同的应用所使用的填充物是不同的。生成的气体在一个恒温色谱柱中分离(例如,5Å分子筛),这一点是非常重要的,由于 N2和CO是同量异位素(质荷比都是m/z 28),并影响H2的电离作用。
尽管主要反应产物为H2、N2和CO,但是一些研究者建议在色谱柱之前使用化学阱来去除微量的其他气体。阱中物质包括:活性炭、高氯酸镁、Sicapent®(附着在黏结剂上 P2O5)和烧碱石棉®。无论H2和CO的相对量如何,使用“吹扫捕获柱”能够让它们接收信号峰的整个基线分离,也能够防止N2干扰 H2同位素测量[29]。
目前,TC/EA-MS联用的分析方法,不仅实现了在线单次分析过程中同时测定微量水的δD和δ18O[24],还可以分析含水矿物和名义上无水矿物中的水含量,同时也可以分析其氢同位素组成。TC/EA-MS联用方法分析样品中水含量和氢同位素组成,其分析下限是样品中的水含量在0.01 L以上。名义上无水矿物中水含量的测定已成为深部地球科学研究的前沿领域[25]。
2 连续流同位素质谱分析系统中的质量控制因素
质量控制是指在分析过程中为保证分析结果真实可靠所采取的任何方法和步骤,它是作为质量保证策略的一部分[30]。以下主要从仪器的外围环境、系统内部参数、样品制备和标准物质的运用等方面进行阐述。
2.1 仪器安装环境的控制
为了获得高精度和高重现性的同位素数据,同位素比值质谱必须置于温度和湿度可控的并能实时监测的环境之中。仪器制造商的预安装和操作培训应该会详细地说明这些参数的允许变化范围。
特别需要提及的是,用于存储同位素比值质谱工作气体(标准气体,准确地说应该是参比气体)的钢瓶(相关的阀门和量表)也要置于温度可控的环境中。因为温度波动会造成工作气体同位素组成较大的漂移,尤其是CO2,其气体状态和其液态流体状态是平衡的[2,16,31]。在 20℃ 温度下,根据饱和数据线可知,CO2工作气体压力低于5.7 MPa要进行更换,因为这一压力表明钢瓶中液态CO2已经不存在。另外,在安全的前提下,工作气体钢瓶应该尽可能靠近仪器。
提供给仪器的载气质量对于数据结果也有影响。几乎所有的连续流配置的载气为氦气,因为氦电离效率低,且对其他气体干扰小。另外,载气进入系统前应该接入过滤器来去除残留的氧气、水汽和碳氢化合物,例如,氦气纯化器(Helium Purifier,VICI,USA)。通常建议在靠近仪器的位置安装一台碳氢过滤器以去除任何痕量的气体,但过滤器不能接在工作气体供应线上,因为它可能引起不可预见的同位素分馏。
2.2 连续流系统参数监测
在开始测量前以及在整个样品分析过程中,确保连续流系统良好的工作状态是非常重要的。实验室建立和遵循一套特定日常仪器检查和质量控制程序,并应用到每一个测量程序。在进行诊断测试和条件实验期间,必须建立起仪器正常测试的规范和标准,这可以确保仪器的可操作性以及在仪器非正常运行时采取相关措施。
2.2.1 气体同位素组成的稳定性(零富集)检验
连续流同位素比值质谱的原始分析结果是相对工作气体的同位素组成进行计算的,因此,在日常分析中监测工作气体同位素组成的稳定性是非常重要的,至少要确保在样品气体流出色谱柱的时间段内工作气体稳定。这种测量对于分析结果的精度和准确度至关重要。
在连续流系统中,稳定性指He携带的工作气体在质谱离子源产生稳定信号。“零富集”(“开-关”)检验就是引入10个工作气体脉冲进入质谱,同时定义一个“标准”脉冲(一般为第二个测量数据,以*标记),其他脉冲相对于“标准”脉冲的δ值的标准偏差来表达。工作气体脉冲的强度应该设置在预期的工作范围内。在色谱-质谱联用分析中,色谱图中会有大量的峰,因此,确定工作气体脉冲合适的出峰时间也很重要。与所有的性能检测一样,一个特定的仪器必须建立认可的评判标准。一般地,对CO2、N2和CO的标准偏差必须小于0.1‰,H2的标准偏差小于1.0‰。
2.2.2 灵敏度和线性调节
仪器用户通常需要对同位素比值质谱进行“灵敏度”或“线性”调节。调节灵敏度来获得最高的信号强度,而调节线性是为在一定的信号强度范围内获得一致的离子比值。与传统的双进样同位素比值分析相比,连续流测量是基于工作气体和样品气之间一次比较,因此更应该注重线性调节。
同位素质谱线性表达为气体同位素组成与离子束强度之间的函数关系,单位为‰/V[32]。经验表明:δ值和峰面积(峰强度)之间的函数关系总是一个线性关系[5,31]。在统计误差范围内,测量的结果会沿着较小斜率直线分布。但是,测试时希望在认可的离子强度范围内的同位素比值与分析物的质量无关或变化很小。通常,在离子强度非常小时会引起非线性效应[5,33],Brand[34]对非线性给出简单的校正方法。另外,Kornfeld等[35]也给出了连续流质谱在较大的动力学范围中的非线性效应校正的方法。
虽然仪器的线性不必进行日常检测,但是必须对工作气体进行定期检测。这种检测与零富集检验类似,不同的是,在这个检测过程中需要将工作气体的离子强度逐步增大。工作气体离子强度范围必须要涵盖待测定样品的离子强度范围,例如,如果样品在3000~8000 mV离子强度范围内进行测量,那么线性测量应该包含的范围是2000~10000 mV。线性范围需要在样品测试之前建立起来。一般情况下,CO2、N2、CO 线性必须小于 0.1‰/V。而2H/1H测量则无需线性检验,因为它需要做日常的系数测定。
我们知道,离子源理想的测试参数很大程度上取决于仪器的类型、离子源的洁净度和许多其他条件[34]。要获得好的灵敏度,需要改变离子源参数来获得工作气体最大的信号强度。要获取好的线性,一些离子源参数要设为“临界”值,例如提取透镜(extraction lens)的参数。然后调节所有的其他参数使工作气体的信号最大。临界值可以通过调节和测量线性的迭代过程来建立,例如,通过设定提取透镜为另一个值并调节所有的其他参数。尽管非常费时,但是这种过程一般仅需要执行一次来建立“临界”值。一些同位素比值质谱软件提供了自动聚焦的功能,这可能加速调节过程,一般在自动聚焦以后再进行手动调节可以获取更好的结果[2]。
2.2.3 背景值监测
在同位素比值质谱测定中,测试背景由两部分构成:一是基线电子漂移;二是当没有气体进入质谱时,相应质量数位置的离子流[34]。背景的处理通常通过质谱的操作软件来完成。标准的方法是在日常测量期间通过首先关闭通向质谱的电磁阀并等候一段时间,抽走质谱中气体来测量背景[34]。
一般情况下,仪器制造商提供的说明书中会详细地说明离子源中可接受的残留气体水平。在实践中,这些背景值在不同的实验室会有所变化,这取决于仪器的配置、使用的气体等级和一些其他因素。重要的是每天都要监测所使用的仪器背景值,这不仅有助于建立起可接受的背景水平,而且同时可以确定其他未知因素对分析结果的影响。
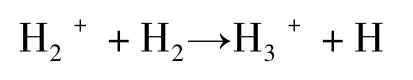
2.3 样品的制备和称量
对于连续流同位素质谱分析方法而言,确保待测试样品、标准物质和质量确认标准(监视标准)以相同方式和相同的处理原则(identical treatment,IT,见下文)来制备和分析是非常重要的[2,3,12,16,26,33-34]。一些化合物要绝对干燥,否则残留的水汽可能影响δ2H和δ18O值。许多化合物都容易吸湿,始终要保证在分析前样品干燥。
EA-IRMS和 TC/EA-IRMS方法都是测定“全样品”同位素比值的方法[2,3,36]。例如,碳同位素比值分析就是燃烧样品中所有的含碳物质。为了使EA-IRMS获得准确的结果,样品要尽可能是均匀分布的。如果某些情况下需要单个化学物种的同位素比值,那么在燃烧/转换过程之前必须对样品进行纯化分离。它可以通过非在线的纯化步骤,或通过结合GC-C-IRMS这样的在线方法来进行。需要特别注意的是,这两种方法可能引起同位素分馏,因此,相同的处理过程也是必要的。
样品称量(使用一台微分析天平)过程也非常重要。合适的样品量可以保证样品生成的CO2和N2(或H2和CO)的信号强度位于同位素比值质谱的线性范围内。理想情况下,来自样品峰的主要离子峰强度应该与工作气主要离子峰强度相匹配。有些情况下可调节工作气体峰强度与样品峰一致[16]。现代的连续流系统还可通过稀释方法使主要离子峰之间尽可能地匹配。标准物质称量一般在100~500 μg之间,通过合适的稀释方法会产生合适的信号强度,所测试样品的称量要依据物质的纯度和含量来确定。
2.4 标准物质的选择和使用
任何一个稳定同位素实验室对标准物质(reference material)选择面临的重要问题有很多。包括:①标准物质的运用:为了使不确定度最小化,应该使用什么样的标准物质?②标准物质δ值范围:这些标准物质需要涵盖自然界中所观测到的δ值范围吗?③标准物质和样品的δ值:它们是否应该具有接近所分析样品的预估δ值?④标准物质使用频次:重复分析标准多次有好处吗?或减少重复分析而使用具有不同的δ值的多个标准物质?⑤标准物质不确定度传递性:当使用多个标准物质时,与标准不确定度相关的误差是如何传递的?
以上这些都是我们进行稳定同位素分析时,使用标准物质所需要解决的问题。
2.4.1 标准物质的运用策略
到目前为止,在稳定同位素测量领域,标准物质的定义还没有一个明确的指南。虽然制定国际通用的计量学基本术语的国际标准机构提供了一致性的一般性定义。但不幸的是,它与稳定同位素测量界建立起来的一些术语是不一致的。广义上“标准”(standard)包括标准物质、已认证标准物质、实验室和内部标准以及标准气体[30]。由于简单的术语“标准”应用于不同目的,是一种模棱两可的表达,它的应用应该受到严格的限制。标准物质是天然的或合成的化合物,它是相对基准标准物质仔细标定过的,属性均匀,具有不确定度。它用来标定实验室装备和分析不同于基准标准物质组成物质测量方法。使用的标准物质应包含具有广泛化学组成和大范围稳定同位素比值[37]。
正如在2.3节所提到的,无论哪种类型的仪器,要做到可靠的稳定同位素分析的基本原则就是遵循样品和标准物质的“同一处理原则”,所谓“同一处理”就是使样品和标准物质经历相同反应路径和反应条件,这是为了考虑发生在质谱分析之前的外部装置中一系列的可能潜在的同位素分馏(例如,不完全转化和气体转移等引起的同位素分馏)。同时,标准物质和样品之间具有类似的组成对于提高分析精度也很重要。正如 Bièvre(1993)等[38]所强调的:如果其他的方面相当,要比较的两个主体越相似,就测量本身而言,产生的误差就越小。这说明了在它们通过外接设备时(EA、TC/EA),要考虑未知样品的化学和物理性质非常重要。然而,对实际样品的同一处理原则的执行又往往并不能如愿:根据定义,需要使用类似和具有一定代表性组成的分析物,而在测试实际自然样品时,往往涉及未知和复杂组成的样品,从严格意义上说,这就使得相同的处理原则很难在实际中应用,但我们可以对标准物质进行处理,使得和样品尽可能地相似。对于一些研究工作中,会缺乏合适的标准物质,这就需要实验室建立自己的实验室标准物质,并评估它们的均一性和稳定性。Zha等[33]对硅酸盐样品中微量碳酸盐分析就采取这样的策略,从而使标准物质具有实际样品类似的成分比例和物质构成,使δ13C和δ18O分析精度和准确度都大幅提高,尤其是使δ18O值受添加磷酸量的影响得以校正。
除了使用标准物质来校准仪器和实验室标准外,标准物质也用来标准化初始数据。选择的标准物质数量和类型会明显地影响标准化,并因此影响最终的结果,有关标准化过程将在数据处理中(第3.3节)来阐述。另外,标准物质还能够用来监测分析方法和同位素比值质谱的性能(所谓的质量保证标准)。可以利用标准物质作为质量控制手段,即:通过对已知同位素组成标准物质的检测,实现在分析过程中不同时间段对系统的质量控制。在分析阶段之前,重复分析工作气体脉冲可检查同位素比值质谱系统的稳定性。
类似地,已认证参考物质(certified reference materials)也可以检测整个分析序列中仪器的稳定性,并监视质量控制方面的精确度。要确保整个分析方法的精度,实验标准物质应该贯穿于分析制备、外接设备和同位素比值质谱分析整个过程。
2.4.2 标准物质的选择
根据Gentile等[30]的研究观点,实验室标准物质应该符合下列条件:①这种物质在制备期间应该很容易处理,并可方便被替代;②应该优先选择高纯度、非吸湿、长时间稳定的化合物;③在室温和大气压下具有低的蒸气压,在高纯溶剂中有好的溶解度(在CSIA);④这种物质的值应该在分析物预期变化的范围内;⑤标准物质应该与分析的样品具有相同或类似的化学组分。
就选择标准物质的δ值接近或远离样品的δ值而言,Skrzypek等[14]分别评估了使用更接近样品δ值和涵盖样品δ值范围的已经标定好的标准物质的优势,在这项研究中,共9个标准物质用于Monte Carlo模型。该模型对所有可能标准物质对之间实现了标准化误差的评估。虽然已经证实了几个优化的标准物质对,但是每一个标准物质对对应一个非常狭隘范围的δ值是优化的。如果仅需要使用两个标准物质,那么其他的16对不同的标准物质被选择涵盖总范围中(从-50‰到 +10‰)很小的部分。虽然这种复杂的选择降低了不确定度,但是在实际应用中不合适,这是由于需要分析多个标准。另外,选择小范围标准物质相关的风险也是非常高的,在这种情况下样品预估的δ值不能总是估计正确的。通过实验,Skrzypek等[14]进一步指出在进行δ13C值分析时,使用一套四个标准物质L-SVEC、NBS-19、USGS24和NBS22(每个分析两次)比16对中任一对优化的小的δ值范围获得的结果有更低的不确定度,结果会更好。因此,分析这四个标准物质几乎涵盖整个δ13C值范围,比任何其他标准物质对更好,即使任何一对标准物质δ值更接近样品的δ值也是这样。对δ18O值分析,Skrzypek等[15]也得出类似的结论。这是由于当标准物质涵盖整个δ值范围的时候不确定度总是更低的。
就每个标准物质测试次数和选择的不同标准物质的数量而言,根据 Skrzypek等[14-15]关于 Monte Carlo模型的统计评估,他们认为只要认真地选择合适的标准物质,那么不确定度会大大降低。对于碳同位素组成分析,如果选择四个标准物质(LSVEC、NBS19、USGS24 和 NBS22),每个标准物质在一批样品中分析两次(总共8个),那么不确定度降低50%。然而,从统计的观点来看,如果仅使用两个标准物质(L-SVEC、NBS19),在每一批样品中分析4次(总共8个),应该会获得类似结果。他们还得出了在EA-IRMS分析的情况下,所谓的组成类似并不是很关键。可以通过同时分析几个不同化学组成的标准物质,并检验仅使用碳酸盐标准或仅使用有机标准物质是否有线性回归方程变化,以此确定不同组成标准物质对的使用对分析结果的影响。根据 Skrzypek 等[13]和 Coplen 等[39]证实,碳酸盐标准物质能够成功地用EA方法进行分析。
使用TC/EA方法分析δ18O的标准化误差与δ13C类似,如果选择两个优化的标准物质组成标准对,且每一个重复分析4次,标准误差近似降低50%[16]。从标准物质和样品化学组成匹配来考虑,在当前可用的已认证标准物质中,使用以下标准物质可以最小化标准化误差:对硝酸盐是USGS35和USGS34,对硫酸盐是IAEA-SO-6和IAEA-SO-5,对有机物质是IAEA-601和IAEA-602。对于所有的标准物质,进行选择的优化原则是涵盖更宽范围的δ值的标准对(包括硝酸盐)。对于IAEASO-6和IAEA-602,它们与标准物质标定相关且有最低的不确定度。然而,这两个标准物质化学组成不同,IAEA-SO-6是一种硫酸盐,IAEA-602是一种有机标准物质。根据其他一些研究,硫酸盐标准物质和有机标准物质一起使用似乎并不会导致化学上组成匹配的问题。对于使用TC/EA方法分析氧同位素的不确定度(近似0.30‰)比EA分析碳同位素(小于0.10‰)的不确定度要高得多。随着样品的δ值偏离程度的增加(相对于δ值为0),不确定度的传递更加明显。因此,样品δ值偏离标准物质对所涵盖的范围时,分析的不确定度会大幅增加[16]。
C和O标准物质已有大量研究。当应用EAIRMS对氮和硫同位素组成分析和TC/EA-IRMS方法对氢同位素组成分析时,由于这三种元素(N、S、H)标准物质的研究程度不如C和O标准物质,因此,关于它们的标准化不确定度传递所知甚少。
总之,在同位素比值质谱分析中,实验室标准物质需要使用较大范围的同位素组成,较理想的情况是能涵盖样品的预期同位素组成。一般工作中至少要使用两个标准物质。标准物质的使用既要考虑样品的性质,同时要涵盖它们未知的同位素比值范围,显然,这是一个挑战。
2.4.3 样品和标准物质序列(sequence)
对于不同的实验室,利用标准物质来标定、标准化和质量控制的策略往往不同。由于标准的操作手册并不能适合所有可能的情况,稳定同位素实验室为了实现最好的分析精度,需要确定标准物质分析数目、类型和同一标准物质分析频次,不同实验室对于样品、标准和空白放置的方法,以及标准之间样品重复样的数量都不尽相同,分析结果不但受到标准物质分析频次的影响,同时标准的数目和它们的位置也能够影响结果。Werner等[26]提出了样品放置列表(序列设计),但是每个实验室都有自己的分析策略。
一般地,好的分析序列需要包括一批样品和一套两个具有不同同位素组成的已知标准物质(理想情况下是两个国际标准物质)。除此之外,分析系统的性能检验必须包括至少一个标准物质或国内标准物质,作为同一批次未知样品的质量控制来进行重复检验。一般情况下,一个批次分析可能包括4个或5个样品(每个样品至少三份)和标准物质(三份),同时标准也用于随后的端元校正(见表1)。根据统计分析原理样品应该分析至少4次(n=4)。如果希望应用概率比,同时控制标准物质(或示例样品)和样品之间可能匹配,那么标准物质和控制标准应该分析偶数多次(例如,n=4,6,…)。很明显,重复的次数越多,则运行的时间越长,周转时间越慢,成本更高。对于氢和氧同位素分析,需要更多次的分析来获得一致值,这是由于一个序列中样品具有不同的同位素组成时会存在记忆效应。在这个序列的开始和结束都要分析每种元素的标准物质,这些值将用来标准化所分析样品的数据。内部质量控制物质在整个分析序列中要定期分析,以确保系统稳定。如果在实验条件下仪器发生漂移,那么样品分析序列中应在其前面或其后面紧跟两个标准物质运行,且每个至少分析三次,以用来进行漂移校正(例如,在样品的批次序列起始和结束之间存在尺度调整,可解释系统差异)。最后,两个空锡杯和银杯应该放在整个样品序列两端,作为空白。有时,在分析样品的过程中也要增加空白次数,这样是为了避免气体色谱柱和水阱饱和引起的背景值增加,同时也可排除来自样品在反应器中的累积污染[8]。
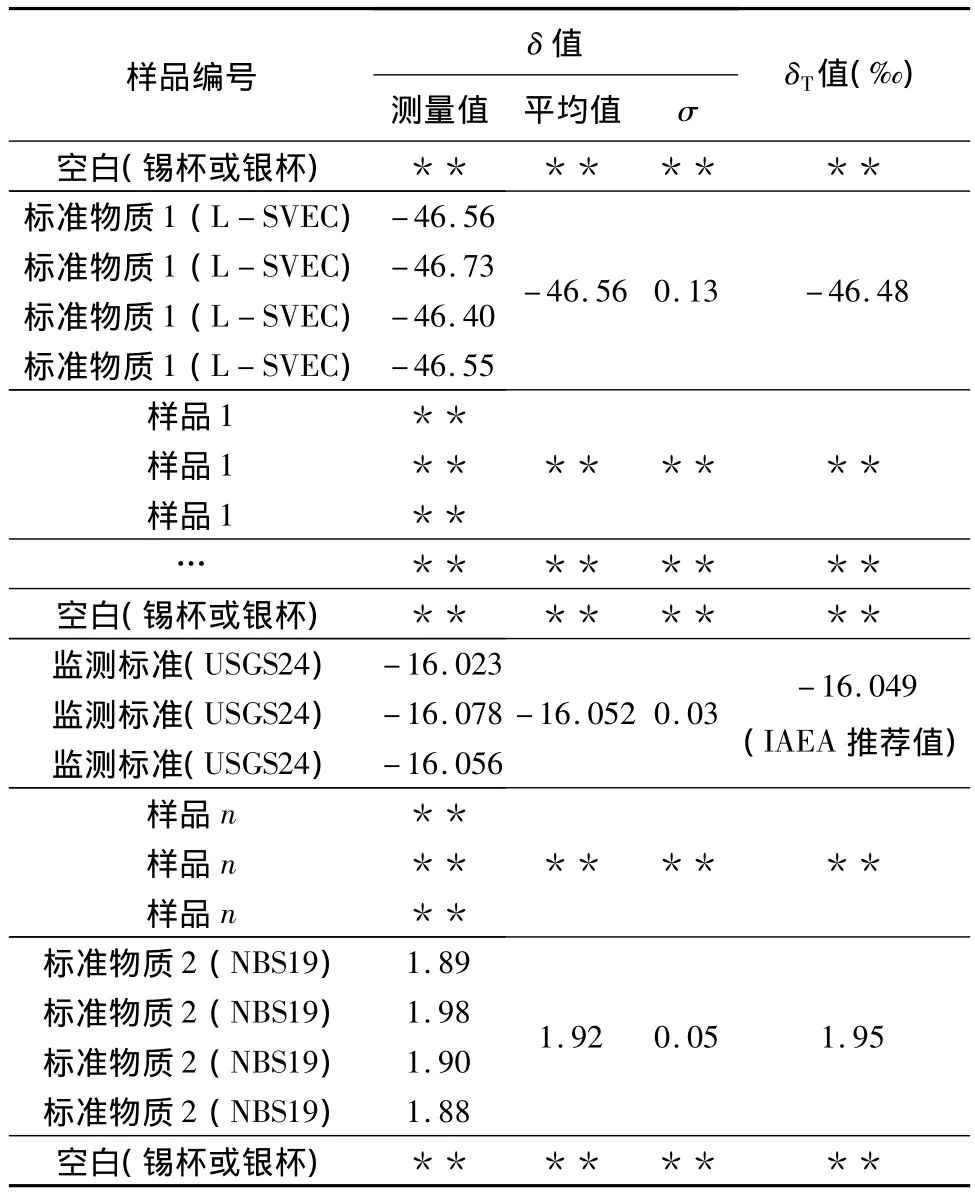
表1 连续流系统条件下的样品和标准物质序列的典型构成(以碳酸盐13C同位素为例)Table 1 The classical sample and reference materials batch sequence compositions in continuous flow experimental conditions(13C isotope analysis of carbonate as an example)
3 同位素数据处理
文献中数据处理多半是一个隐蔽过程。毫无疑问,恰当地数据处理会影响最终的分析结果。原始数据的处理一般包括质量控制过程监测、结果的保留和舍弃策略(样品重复分析)以及原始数据对国际参比尺度的标准化(也称为校正或转化)、不确定度计算等。
3.1 同位素校准(calibration)
根据通用计量术语及定义指出同位素比值校准过程(calibration)包括两步:第一步是建立国际标准(或已认证同位素参比物质,或同位素重量分析物)的同位素比值与同位素质谱的离子数之间的函数关系。第二步是使用这种函数关系来建立未知样品的离子流测量值或离子数与样品的原始同位素比值之间的关系。校准过程可以通过校准方程、校准图、校准曲线或校准表来实现[40]。实际上,原始数据的标准化过程也是校准过程一个环节,两个国际标准校准过程是多点线性标准化的一个特例。
3.2 控制曲线
控制曲线是内部数据质量检查的有力工具,它记录所有分析数据同时监测样品分析过程是否可控,对此Werner等[26]已有详细的报道。通过监测分析结果的平均值和分散度,控制曲线可检查这些分析过程和分析结果是否对时间、不同批次、不同天或不同月产生响应。这种对系统长期性能观察,本质上就是防止产生错误的结果,可实现对分析偏差是趋势性或是阶段性的增加和降低的判断。平均值(曲)线为最常见的控制(曲)线,表达为数据生成的曲线是围绕着已知值(为几次测量的平均值或认证标准物质的已知同位素值)的中心线,并带有由用户自己定义的起警示作用的上下限(警戒限)。通常,警戒限和作用线的标准平均偏差为2σ和3σ,在平均值(曲)线所有值中分别覆盖有95.5%和99.7%的置信区间[2]。当一个点位于作用线的外部或两个连续的点位于警戒限的外部,我们应该采取措施来恢复过程为可控状态。图2为2013年中国科学院壳幔物质与环境重点实验室内部监测标准(04BXL07)的控制曲线图。从图2可以看出,虽然这种标准所测量的点几乎都落在2σ内,但在2013年5月之前值偏低一些,2013年6月有向上漂移倾向。通过对这些数据进行拟合,线性相关性较差,说明了系统随时间和不同批次有变化。从分散度来看,2013年5月之后,数据散布更大些。
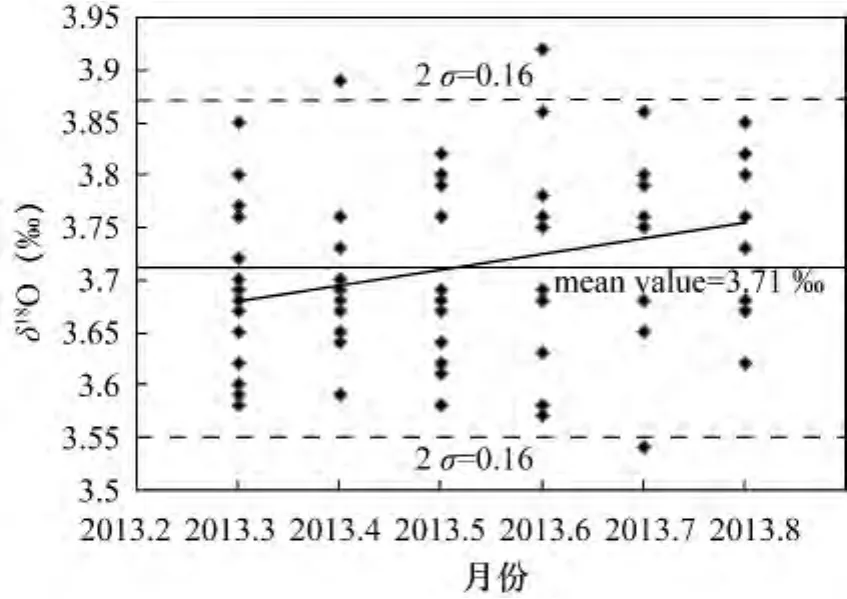
图2 内部监测标准物质04BXL07的δ18O质量控制曲线图Fig.2 A quality control chart for the determination of δ18O using in-house monitoring standard 04BXL07
3.3 标准化过程(normalization)
所有的现代化连续流同位素质谱都装备有一个让相应的工作气体无质量歧视进入到离子源的入口(inlet)。尽管这种防护措施不能防止质谱性能变化,但可以监控或者修正由此带来的变化,可还不能彻底解决例如样品和标准物质通过“相同处理原则”对分析结果的标准化这样的重大问题[26]。
同位素质谱是高精度而非高准确度的一种仪器[16],且稳定同位素组成不是作为绝对比值测量,而是相对于一个标准物质来测量的。在实际同位素测量中,这个内部的标准物质通常是一种实验室标准气体(工作气体),它是同样品气交替测量多次(双路进样系统),或在样品之前或之后测量多次(例如,ThermoFisher公司的conflo仪器)。或者,每隔几个样品分析一个固体标准物质(例如,Sercon20-20)。不同实验室由于运用不同的已认证的或标定的参比气体(标准气体,reference gas,working gas)以及标准物质,因此获得的结果是与实验室使用的标准有关的原始值,需要通过一个国际尺度(例如,VSMOW、VPDB、VCDT或空气)转化样品原始(测量)的δ值为报道的“真实”的δ值,从而保证不同的实验室所获数据具有可比性。数学上重新计算的过程通常称为标准化。这个过程的一些方面在同位素比值质谱软件内部自动完成的,对分析者而言可能不可见。补充的标准化可能要在外部电子文档中进行(两点或多点标准化过程)。
据文献[2,12,16]报道,初始数据标准化为国际参比尺度能够采取的方式:①相对于已知同位素比值的参比气体(此步骤与相同处理原则相违背);②相对单一的参比物质;③相对于两个或多个参比物质,这种方法常常将标准化和校准过程很好地统一起来。正如Paul等[12]所述,使用不同的标准化方法可能导致重大的差别,仅由于不同的数学算法,其结果在不同的实验室间甚至分析相同的样品获得的δ值有重大的差异。
一般的,同位素比值质谱软件相对工作气体自动计算同位素比值,被称为“单点锚定”(singlepoint anchor)计算方法。这需要操作者指定工作气体的真实的 δ值[δT(wg)=[(Rwg/Rstd)-1]×1000]。工作气体和样品原始的δ值被测量后,样品的“真实”δ值以方程(1)表示:
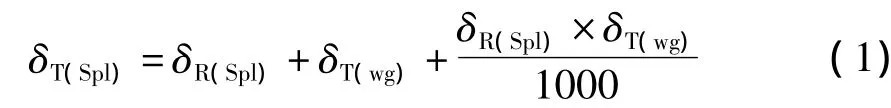
式中,δT(Spl)表示样品的真实δ值,δR(Spl)表示样品相对工作气体的原始值,δT(wg)表示工作气体的真值。
为了减小不确定度,样品的同位素组成必须接近工作气的同位素组成。标准化的不确定度随着[δT(Spl)- δT(wg)]增加而增大[12,16]。另外,这种标准化是要求钢瓶中(对于Conflo系统,通常装在高压钢瓶中)标准气体消耗完之前,或至少在下一次标定之前一直有一个恒定的δ值为基础的。由于工作气体的组成存在质量相关分馏,即工作气体同位素组成会随着时间发生变化(一般轻同位素会变得更轻,重同位素会变得更重),因此对来自高压钢瓶中工作气体δT(wg)定期进行标定测量是非常重要的。
单点锚定也可相对一种固体标准物质进行。与相对于钢瓶气标准化相比的优点是样品和标准都经历了相同条件下的化学处理过程而生成的分析气体。但如果仅仅使用一个标准,意味着这种标准化方法中的不确定度会相对于样品和标准相互之间的δ值差值成比例增加。样品和标准物质的原始δ值是相对工作气体测量,然后根据方程(2)来计算样品的真实的δ值。
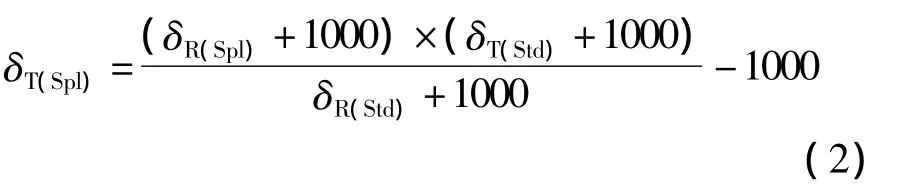
式中,δT(Spl)表示样品的真实δ值,δR(Spl)表示样品的原始δ值,δT(Std)表示标准的真实δ值,δR(Std)表示标准的原始δ值。
以上两种标准化都是单锚点标准化,对于这样的单点锚定,如果没有严格地遵守同一处理原则,并且标准和样品的同位素差异较大,那么这种方法将产生较大的标准化不确定度。
第三种方法是基于线性回归的标准化,截距和斜率用于对原始数据的同位素度量尺度的重新计算。就标准物质的数量而言,称为两点或多点标准化方法。校准线或校准曲线并不是一种新方法,这种方法常用在化学分析中进行标准化。然而,这种方法对于稳定同位素分析而言相对是一种新应用。最初,在建立了VSMOW曲线常用度量尺度后,两点方法用于水的氢和氧稳定同位素组成测量的标准化。后来,Coplen等[39]使用 NBS19和 L-SVEC建立了VPDB标准度量尺度。
Paul等[12]和 Skrzypek[16]的方法至少用两个不同标准物质的原始测量δ值作为横坐标(x轴),真值或通常称为可接受的δ值,作为纵坐标(y轴,图3)。这些点构成了一条回归线(图4)跨过不同的δ值范围,取决于分析的标准的δ值:

式中,a表示斜率,b表示截距。
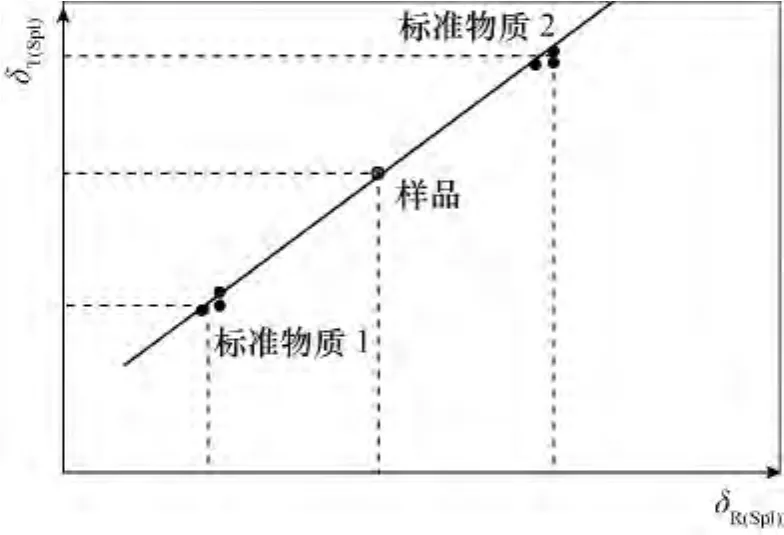
图3 样品对标准的两点标准化示意图Fig.3 The diagrammatic sketch of a two-point normalization method using linear regression based on two wellcalibrated reference materials,recalculating sample raw value to international scale
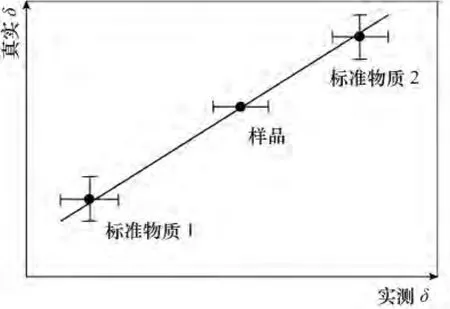
图4 样品的合成不确定度示意图(据文献[2]修改)Fig.4 The diagrammatic sketch of combined uncertainty of sample(revised by Reference[2])
在一段时间内,回归线斜率a近似为常数1(1.00±0.05)。如果使用同一套标准,斜率的变化可能反映分析的不确定度,即测量期间条件的改变或仪器背景值的改变。截距b主要与系统中使用的工作气体的δ值有关(方程3)。样品的标准化过程为样品的原始的δ值(对工作气体或固体标准)乘上一个斜率然后加上一个截距值。因此,截距对所有要标准化的样品都是相等的,与样品的δ值无关。对于斜率而言,当样品的δ值远离零值时,斜率的影响会进一步增加。
使用超过两点线性标准化的方法,也称作为“多点线性标准化”或简称为“校准曲线”,这可能是基于拟合方法的最好的回归线。系数R2表明这些测量数据遵守线性关系的紧密程度(判定观察值近似均匀的分布),以及标准物质测量值的随机不确定度效应(例如,不完全燃烧)。
3.4 样品不确定度计算
不确定度是指由于测量误差的存在,对被测量值与其真值之间差异的无法确定性。当前同位素比值质谱能够测定同位素比值的不确定度一般要好于0.02‰。对于氢而言,不确定度通常要大一个数量级,这是因为自然界中2H/1H同位素比值要比其他元素的同位素比值要小几个数量级。大的不确定度一般是在同位素比值质谱分析之前样品的前处理过程中产生的。
当测量结果是由若干个其他测量值求得时,按其他测量值的方差和协方差计算的标准不确定度,称为合成标准不确定度[41]。它是测量结果标准偏差的估计值,用符号UC表示。根据文献[2]报道的δ值合成不确定度可能来自:(a)重复样测量的精度;(b)实验过程偏差;(c)用于标准化δ度量尺度的标准物质的δ值不确定度;(d)校正和标准化数据所采用的算法。通过选择合适的分析条件和运用IT原则,(a)和(b)都能够减小。而对于(c)和(d)而言,同一实验室内部测量结果类似,各个实验室间的不确定度会随着样品和参比物质之间δ值差异增加而增加。这表明了(c)和(d)对于测定结果的不确定度有重大的影响。实验室内部无法处理关于标定物质和实验室间对比物质(对于组成均一性物质而言)相关的不确定度。虽然这些不确定度随着时间累积将渐渐减小,但是仍然必须结合不确定度预计值和实验室内部标准对这些标准物质标定所产生相关的不确定度有所预计。
合成不确定度可以用图4表示。垂直误差棒表示用于校准/标准化物质的δ值不确定度,水平误差棒表示任意物质原始测量的δ值不确定度。样品δ测量值不确定度用中间的水平误差棒表示。
为了能够对结合不同的不确定度贡献的某一具体的样品所测结果给出唯一的不确定度估计,不确定度计算必须是采用相同的数学表达方式。根据国际认可的不确定度计算规则,不确定度应该以标准偏差来表示。对合成的不确定度基本规则是“平方和的平方根规则”。以标准偏差表示的不确定度成分Ux1,…,Uxn按照方程(4)合成,给出最终结果的不确定度UC:
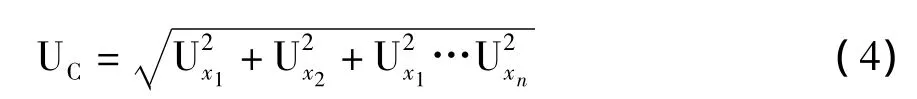
然而,这需要测量结果中不确定度组分要采用相同的单位(例如,就同位素比值质谱测量而言以δ值表示),且不确定度组分之间要不相关。
如果使用两个标准物质Std1和Std2的测量值和真值进行标准化,那么计算δT(Spl)的方程:
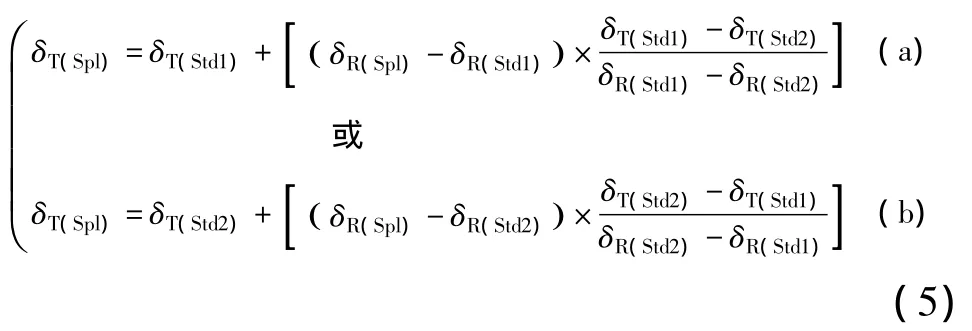
现就式(5)中的(a)展开讨论。
由于 δT(Std1)和 δR(Std1)项在方程(5a)中出现两次,所以合成不确定度不能使用简单的规则。最为直接的途径按照Kragten方法描述的基于电子数据表的计算[41]。以 δ2HT(Spl)中不确定度计算作为实例,电子数据表形式如表2所示。通过分析VSMOW和SLAP进行标准化。以上标准的δ2H真值的标准偏差在不同情况下由IAEA给定是0.3‰。由于随机不确定度,δR(VSMOW)和δR(SLAP)标准不确定度是1.0‰(重复测量平均值的标准偏差)。样品的δR(Spl)值为-120.0‰,标准差为1.2‰(重复测量平均值的标准偏差)。
需要计算结果的参数值和相关的标准不确定度都列在表2所示的B列和C列。用来计算结果的公式都列在单元格B7中。B列中数据拷贝到D~H列(在计算δT(Spl)时使用每一列中一个参数)。在单元格C2中所给的不确定度添加到单元格D2中,单元格C3中不确定度添加到单元格E3中,等等。单元格D7到H7显示了对δT(Spl)重新计算的值,包括单个参数中不确定度效应。第8行给出了重新计算的值和在单元格B7中对δT(Spl)最初计算之间的差值,显然,很容易看出不同的参数对δT(Spl)重新计算的不确定度影响的大小。δT(Spl)标准偏差(单元格C7)是通过对第8行差值的平方,再加和,然后平方根获得的。
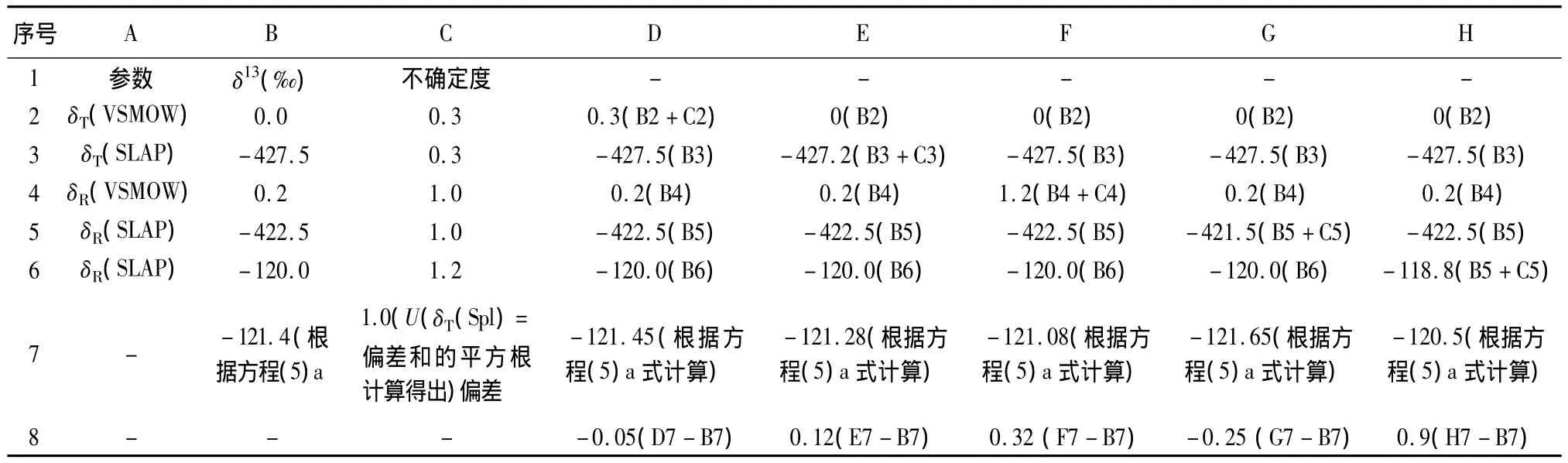
表2 使用合成标准不确定度来计算样品的不确定度Table 2 Calculation of uncertainty of sample utilizing combined standard uncertainty
3.5 数据的保留和舍弃
样品数据的保留和舍弃应该是与质量控制策略相一致。观测到异常结果就必须要对分析的有效性产生疑问,或许要尝试着重新分析样品,但前提是要有足够的样品可用。而且,所有的样品应该做相同的处理。仅当质量控制标准证实了分析问题所在,并且周围环境已被证实再次分析是合理的情况下,样品才应该进行重新分析。
4 结语
连续流同位素质谱系统(EA-IRMS和TC/EA-IRMS)具有便捷、快速、样品用量少以及不需要前处理或很少前处理的优势,特别是原位同位素分析的发展,在今后的稳定同位素分析当中,连续流质谱方法显然是不可或缺的中心角色,在实际应用中也会越来越广泛。随着同位素比值分析连续流系统覆盖范围扩展,应用于测量稳定同位素比值越来越广泛,这就要求最好运用好的工作实践知识,来确保仪器的稳定,以及好的同位素质谱工作知识,来维持在日常分析中高精度的分析能力。因此,同位素分析者严格遵守一套完备的操作方案和质量控制协议,以及掌握合理的数据处理方法对获得高精度和准确的同位素比值,并能够和其他的实验室获得一致的同位素数据是大有裨益的,且显得尤为迫切。在同位素分析中主要需做到以下几点。
(1)稳定同位素实验室需要建立和遵循一套特定日常仪器检查和质量控制,并在开始测量和整个样品分析过程中确保系统良好的工作状态。分析之后,如发现标准物质结果超过性能控制曲线警戒限制,应该采取措施来恢复过程为可控状态。
(2)基于线性回归的两点或多点标准化方法是最好的校准方法。只要可能,应该要做到基于标准物质的标准化线性回归,这些标准和同批次样品一起运行,执行同一处理原则,且它们的δ值应该涵盖自然界中所观察到的自然丰度的整个变化范围,且需要长期连续使用同一套标准,样品和标准序列也应合理地安排并且要统一。
(3)在数据处理中要使用合成标准不确定度来确定样品的不确定度。
[1]Midwood A J,McGaw B A.Recent developments in the analysis of light isotopes by continuous flow isotope ratio mass spectrometry[J].Analalytical Communication,1999,36:291-294.
[2]Carter J F,Barwick V J.Good Practice Guide for Isotope Ratio Mass Spectrometry,FIRMS[M].ISBN 978-0-948926-31-0.2011.
[3]Brenna J T,Corso T N,Tobias H J,Caimi R J.Highprecision continuous-flow isotope ratio mass spectrometry[J].Mass Spectrometry Review,1997,16:227-258.
[4]Révész K M,Landwehr J M.δ13C and δ18O isotopic composition of CaCO3measured by continuous flow isotope ratio mass spectrometry:Statistical evaluation and verification by application to Devils Hole core DH-11 Calcite[J].Rapid communications in Mass Spectrometry,2002,16:2102-2114.
[5]Spötl C,Vennemann T W.Continuous-flow isotope ratio mass spectrometric analysis of carbonate minerals[J].Rapid Communications in Mass Spectrometry,2003,17:1004-1006.
[6]Fritzsche F,Tichomirowa M.Signal improvement in elementalanalyzer/continuous flow isotope ratio mass spectrometry for samples with low sulfur content using a pre-concentration technique foron-line concentration adjustment[J].Rapid Communications in Mass Spectrometry,2006,20:1682-1697.
[7]Grassineau N V,Mattey D P,Lowry D.Sulfur isotope analysis of sulfide and sulfate minerals by continuous flow-isotope ratio mass spectrometry[J].Analytical Chemistry,2001,73(2):220-225.
[8]Grassineau N V.High-precision EA-IRMS analysis of S and C isotopes in geological materials[J].Applied Geochemistry,2006,21:756-765.
[9]Fry B,Silva S R,Kendall C,Anderson R K.Oxygen isotope corrections for online δ34S analysis[J].Rapid Communications in Mass Spectrometry,2002,16:854-858.
[10]Yun M,Mayer B,Taylor S W.δ34S measurement on organic materials by continuous flow isotope ratio mass spectrometry[J].Rapid Communicationsin Mass Spectrometry,2005,19:1429-1436.
[11]Fry B.Coupled N,C and S stable isotope measurements using a dual-column gas chromatography system[J].Rapid Communications in Mass Spectrometry,2007,21:750-756.
[12]Paul D,Skrzypek G,Forizs I.Normalization of measured stable isotope composition to isotope reference scale—A review[J].Rapid Communications in Mass Spectrometry,2007,21:3006-3014.
[13]Skrzypek G,Paul D.δ13C analyses of calcium carbonate:Comparison between the Gasbench and elemental analyzer techniques[J].Rapid Communications in Mass Spectrometry,2006,20:2915-2920.
[14]Skrzypek G,Sadler R,Paul D.Error propagation in normalization of stable isotope data:A Monte Carlo analysis[J].Rapid Communications in Mass Spectrometry,2010,24(18):2697-2705.
[15]Skrzypek G,Sadler R.A strategy for selection of reference materials in stable oxygen isotope analyses of solid materials[J].Rapid Communications in Mass Spectrometry,2011,25:1625-1630.
[16]Skrzypek G.Normalization procedures and reference material selection in stable HCNOS isotope analyses:A review[J].Analytical and Bioanalytical Chemistry,2013,405:2815-2823.
[17]王政,刘卫国,文启彬.土壤样品中的氮同位素组成的元素分析仪-同位素质谱分析方法[J].质谱学报,2005,26(2):71-75.
[18]曹建平,黄奕普,刘广山,陈敏,李鸿宾.海洋悬浮颗粒中氮同位素的EA-IRMS法测定[J].台湾海峡,2003,22(1):1-8.
[19]崔杰华,祁彪,王颜红.植物样品中稳定碳同位素的EA-IRMS系统分析方法[J].质谱学报,2008,29(1):24-29.
[20]王旭,张福松,丁仲礼.EA-Conflo-IRMS联机系统的燃烧转化率漂移及其对氮碳同位素比值测定的影响[J].质谱学报,2006,27(2):104-109.
[21]储雪蕾.一种新的、快速的碳、氮、硫同位素测定手段——EA-IRMS连线分析技术[J].矿物岩石地球化学通报,1996,15(4):259-262.
[22]郑永飞,龚冰,王峥荣.岩石中的碳同位素比值的EA-MS测定及其地球化学应用[J].地质论评,1999,45(5):529-538.
[23]张媛媛,贺行良,孙书文,朱志刚.元素分析仪-同位素比值质谱仪测定海洋沉积物有机碳稳定同位素方法初探[J].岩矿测试,2012,31(4):627-631.
[24]刘运德,甘义群,余婷婷,刘存富,周爱国.微量水氢氧同位素在线同时测试技术——热转换元素分析同位素比质谱法[J].岩矿测试,2010,29(6):643-647.
[25]龚冰,陈仁旭,郑永飞.大别—苏鲁造山带超高压变质岩矿物水含量和氢同位素组成[J].科学通报,2013,58(22):2169-2174.
[26]Werner R A,Brand W A.Referencing strategies and techniques in stable isotope ratio analysis[J].Rapid Communications in Mass Spectrometry,2001,15:501-519.
[27]Preston T,Owens N J P.Interfacing and automatic elemental analyser with an isotope ratio mass spectrometer:The potential for fully automated total nitrogen and nitrogen-15 analysis[J].Analyst,1983,108:971-977.
[28]Glesemann A,Jäger H J,Norman A L,Krouse H R,Brand W A.On-line sulfur isotope determination using an element analyser coupled to a mass spectrometer[J].Analytical Chemistry,1994,66:2816-2819.
[29]Fourel F,Martineau F,Lécuyer C,Kupka H J,Lange L,Ojeimi C,Seed M.18O/16O ratio measurement of inorganic and organic materials by elemental analysispyrolysis-isotope ratio mass spectrometry continuous flow techniques[J].Rapid Communications in Mass Spectrometry,2011,25:2691-2696.
[30]Gentile N,Besson L,Pazos D,Delémont O,Esseiva P.On the use of IRMS in forensic science:Proposal for a methodologicalapproach[J].Forensic Science International,2011,212:260-271.
[31]Finnigan Gasbench Ⅱ Operating Manual[Z].
[32]Nelson S T.Sample vial influence on the accuracy and precision of carbon and oxygen isotope ratio analysis in continuous flow mass spectrometric applications[J].Rapid Communications in Mass Spectrometry,2000,14:293-297.
[33]Zha X P,Zhao Y Y,Zheng Y F.An online method combining a GasbenchⅡwith continuous flow isotope ratio mass spectrometry to determine the content and isotopic compositions of minor amounts of carbonate in silicate rocks[J].Rapid Communications in Mass Spectrometry,2010,24:2217-2216.
[34]Brand W A.Mass Spectrometry Handware for Analyzing Stable Isotope Ratios[M]//de Groot P A,eds.Handbook ofStableIsotope AnalyticalTechniques(Vo1.1).Oxford:Elsevier B V,2004:805-819.
[35]Kornfeld A,Horton T W,Yakir D,Turnbull M H.Correcting for nonlinearity effect of continuous flow isotope ratio mass spectrometry across a wide dynamic range[J].Rapid Communications in Mass Spectrometry,2012,26:460-468.
[36]Muccio Z,Jackson G P.Isotope ratio mass spectrometry[J].Analyst,2009,134:213-222.
[37]Gröning M.International Stable Isotope Reference Materials[M]//de Groot P A,eds.Handbook of Stable Isotope AnalyticalTechniques(Vo1.1).Oxford:Elsevier B V,2004:874-906.
[38]Bièvre P D,Laeter D,Peiser H S,Reed W P.Reference materials by isotope ratio mass spectrometry[J].Mass Spectrometry Review,1993,12:143-172.
[39]Coplen T B,Brand W A,Gehre M,Gröning M,Meijer H A J,Toman B,Verkouteren R M.New guidelines for δ13C measurement[J].Analytical Chemistry,2006,78:2439-2441.
[40]Coplen T B.Guidelines and recommended terms for expression of stable-isotope-ratio and gas ratio measurement results[J].Rapid Communications in Mass Spectrometry,2011,25:2538-2560.
[41]中国合格评定国家认可委员会.化学分析中的不确定度的评估指南[S].2006:6.
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
同位素(2018年1期)2018-01-18
河北遥感(2017年2期)2017-08-07
中学生数理化·教与学(2017年4期)2017-04-22
中国医学影像学杂志(2015年9期)2015-12-15
同位素(2014年3期)2014-06-13
同位素(2014年2期)2014-04-16
同位素(2014年2期)2014-04-16
特产研究(2014年4期)2014-04-10
中国环境科学(2014年4期)2014-02-02
