
荧光定量PCR检测重组新蛭素中毕赤酵母基因组DNA的残留量
2014-10-27 09:04:44刘晶晶郭莹莹李艳琪王文文吴祖泽靳继德
生物技术通讯 2014年3期
刘晶晶,郭莹莹,李艳琪,王文文,吴祖泽,靳继德
1.北京工业大学 生命科学与生物工程学院,北京 100022;2.军事医学科学院 放射与辐射医学研究所,北京 100850;3.天津大学 化工学院,天津 300072
毕赤酵母不但具有繁殖快、易培养、培养基廉价和试验过程简单可行等优点,而且具有强启动子、可以对外源蛋白进行加工折叠和翻译后修饰的特点,因此具备了典型的真核生物表达体系的条件。毕赤酵母已发展成一个较为理想的真核蛋白表达系统,在国内外被广泛应用于生产外源蛋白和生物制品,已有1000多种外源蛋白在毕赤酵母中获得表达[1]。目前,生物制品的生产大都借助原核或真核表达系统,但表达系统宿主又会带来一定的安全性问题,因此这些生物制品必须符合严格的质量标准[2-3]。宿主菌残留的DNA是此类产品中特有的潜在致癌性杂质,这些产品中外源性DNA残留量的控制是一个非常重要的指标[4-5]。《中华人民共和国药典》对生物制品中宿主DNA的残留量有明确规定,即原核表达系统生产的产品,DNA残留量标准是每人份不超过10 ng,真核细胞生产的产品其含量应不高于每人份100 pg,酵母表达系统生产的产品其含量不高于每人份10 ng[6]。酵母属真核生物,所以应制定更为严格的DNA残留量质量控制标准,以提高产品的安全性。药典推荐使用的外源DNA残留检测方法有斑点杂交法和荧光法。DNA杂交法实验操作周期长,步骤烦琐,干扰因素多,重复性差,且不能进行定量分析[7-8],一次检测的样本量也比较有限。荧光法的检测灵敏度较低,检测范围为1~80 ng/mL。此外,针对DNA重组药物中残留的宿主DNA含量的测定方法还有基于DNA结合蛋白的Threshold Immuno⁃assay分析系统[9]及实时定量PCR法[10]。实时定量PCR(real-time PCR)技术自从1993年问世以来,就以其准确、灵敏度高、便捷、低污染等特点而被广大科研工作者广泛应用于基因表达分析、病原物和转基因产品检测等方面[11-13]。它既保持了PCR技术灵敏、快速的特点,又克服了以往PCR技术中存在的假阳性污染和不能进行准确定量的缺点,还具有重复性好、省力、低费用等优点[14]。在本研究中,我们建立了基于SYBR GreenⅠ荧光染料的实时定量PCR检测方法,实现了对重组产品中毕赤酵母DNA残留量的快速、简便、高灵敏度的定量检测,可用于重组新蛭素等生物制品制备过程中的质量控制。
1 材料与方法
1.1 材料
毕赤酵母菌GS115为本室保存;5批注射用重组新蛭素为本实验室制备,均为毕赤酵母菌GS115表达生产的生物制品。SYBR Premix Ex Taq(Perfect Real Time)购自大连TaKaRa公司;PrepSEQ残留DNA提取试剂盒购自北京东方嘉诚科贸有限公司;酵母基因组提取试剂盒购自Promega公司;溶细胞酶(lyticase)购自天根生化科技公司;全自动荧光定量PCR_LightCycler480系统为Roche公司产品。
1.2 检测基因与扩增引物
选择毕赤酵母基因组中拷贝数高、染色体中分布广的5S rRNA基因作为扩增的目的基因,根据re⁃al-time PCR引物设计原则设计特异性扩增引物Primer 5S-sense(5'-GGTTGCGGCCATATCTAG-3')和 Primer 5S-antisense(5'-AGATTGCAGCACCTGA GT-3')[15],由北京奥科鼎盛生物科技有限公司合成。
1.3 宿主菌基因组DNA标准品的制备
取毕赤酵母500 μL接种于100 mL YPD培养基中,30℃振荡培养至D600nm约为1.5,离心收集菌体,加入山梨醇缓冲液(0.12 mol山梨醇,用pH7.4的0.1 mol/L磷酸钠缓冲液定容至100 mL)300 μL、溶细胞酶(10 U/μL)10 μL,37℃摇床温育60 min,冷却至室温,离心弃上清,用酵母基因组提取试剂盒提取宿主菌DNA,分别用异丙醇沉淀和70%乙醇洗涤去掉酵母基因组DNA中的脂溶性物质,最终得到的沉淀加入50 μL无菌水和1.5 μL RNase,37℃温育15 min,65℃温育1 h或4℃过夜,得到酵母基因组DNA,保存在-20℃,用紫外分光光度法测定浓度及D260nm/D280nm值。
1.4 real-time PCR检测毕赤酵母DNA残留量方法的建立
将基因组DNA稀释成不同浓度的标准品(1000、100、10、1、1×10-1、1×10-2、1×10-3、1×10-4pg/μL),取10 μL基因组DNA标准品为模板,以Prim⁃er 5S-sense和Primer 5S-antisense为引物,在全自动荧光定量PCR_LightCycler480系统中进行扩增,实时检测荧光强度。real-time PCR反应体系包括SYBR Premix Ex Taq(Perfect Real Time)(2 ×)12.5 μL、引物(10 μmol/L)各 0.5 μL、基因组 DNA标准品 10 μL、无菌 ddH2O 1.5 μL(DNA 聚合酶、PCR缓冲液、dNTP及SYBR GreenⅠ均已事先混合于商品化的SYBR Premix Ex Taq中)。经过优化的反应条件为:95℃预变性5 min激活Taq聚合酶,95℃变性5 s、55℃退火15 s、72℃延伸15 s,循环70次,55~95℃程序升温检测熔解曲线,进行荧光检测。
1.5 精密度和回收率测定
将1000 pg/μL的标准品DNA用灭菌注射用水稀释至1 pg/μL,平行做8孔,进行标准品的精密度测定;同时取供试样品稀释至20 mg/mL,平行做3孔,测定样品的精密度。为了测定DNA标准品在样品中的加样回收率,先用灭菌注射用水将样品溶液稀释至20 mg/mL,然后将标准品DNA分别稀释至1、1×10-1和1×10-2pg/μL,分别取5 μL标准DNA再加5 μL样品溶液作为待测样品1、2和3,每个浓度平行加样3孔,进行PCR反应,测定加样回收率。用全自动荧光定量PCR_LightCycler480系统进行数据分析结果。用下式计算DNA加样回收率。为了减少待测样品中的高浓度蛋白对PCR结果的干扰,应用DNA纯化回收试剂盒对DNA样品进行回收,观察回收对DNA含量测定的影响。
1.6 重组新蛭素制品中DNA含量的测定
根据每人次用剂量(20 mg),将所有供试样品均稀释到20 mg/mL进行测定,再利用回收率校正计算样品DNA残留量,用下式计算(x代表标准曲线的横坐标值,即样品浓度的对数)。
2 结果
2.1 宿主菌基因组DNA标准品的制备结果
利用紫外可见分光光度计检测酵母基因组DNA,以去离子水为空白对照,测得DNA浓度为527 μg/mL,D260nm/D280nm值为1.824。
2.2 real-time PCR定量检测标准曲线的建立
图1为1×10-4~1000 pg/μL的标准品DNA realtime PCR扩增曲线。可以看出1×10-1~1000 pg/μL标准品的扩增曲线平稳光滑,峰值较高,没有杂合的基因片段,且指数增长期、线性增长期及平台期呈现明显的“S”型曲线,标准品DNA扩增效率较高。而标准品低于1×10-2pg/μL时,虽然也可获得较好的扩增曲线,但重复性和扩增效率明显降低,而且当选取DNA浓度为1×10-2~1000 pg/μL制作标准曲线,所得标准曲线的误差值为0.271,大于0.2;选取DNA浓度为1×10-1~1000 pg/μL制作标准曲线,所得标准曲线的误差值为0.197。标准曲线的斜率为-5.055,截距为29.85,扩增效率为1.577。图2为实时荧光定量PCR的熔解曲线,可以看出DNA浓度为1×10-1~1000 pg/μL时可检测到单一明显的特异峰值,且无非特异性扩增杂峰及引物二聚体的低矮小峰出现,证明了产物的特异性。当浓度低于1×10-2pg/μL时,熔解曲线明显变得低矮,且形态发生了变异。PCR结束后,用PCR_LightCycler480系统进行数据分析,图3为标准曲线。样品浓度与循环阈值(Cp)之间的线性关系曲线表达式为Cp=-5.055x+29.85(x是标准品浓度取对数后的值),从仪器读取待测样品的Cp值,然后把待测样品的Cp值代入线性方程,就可以得到x值,算出其初始浓度。

图1 real-time PCR扩增曲线

图2 熔解曲线
2.3 精密度测定结果
DNA标准品含量为1 pg/μL时的精密度测定结果见表1,Cp值的RSD为3.09%,标准品测定结果平均值为0.71 pg/μL,RSD为48.39%。测定供试样品DNA残留量精密度结果见表2,平均值为2.7×10-4pg/μL,RSD为25.0%。
2.4 回收率测定结果
毕赤酵母DNA加样回收率测定结果见表3,标准品浓度为1×10-2~1 pg/μL时的回收率为32.4%~80.0%。对浓度分别为100、10和1 pg/μL的标准品经DNA纯化试剂盒纯化后进行回收率测定,结果见表4,标准品经过纯化后的回收率较低,原因可能是部分DNA在纯化操作过程中损失了。
2.5 供试样品DNA含量测定结果
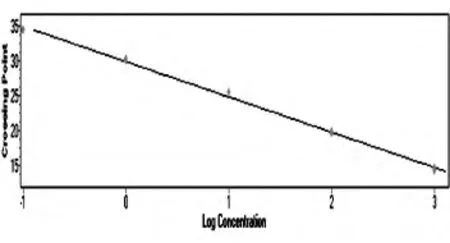
图3 real-time PCR标准曲线
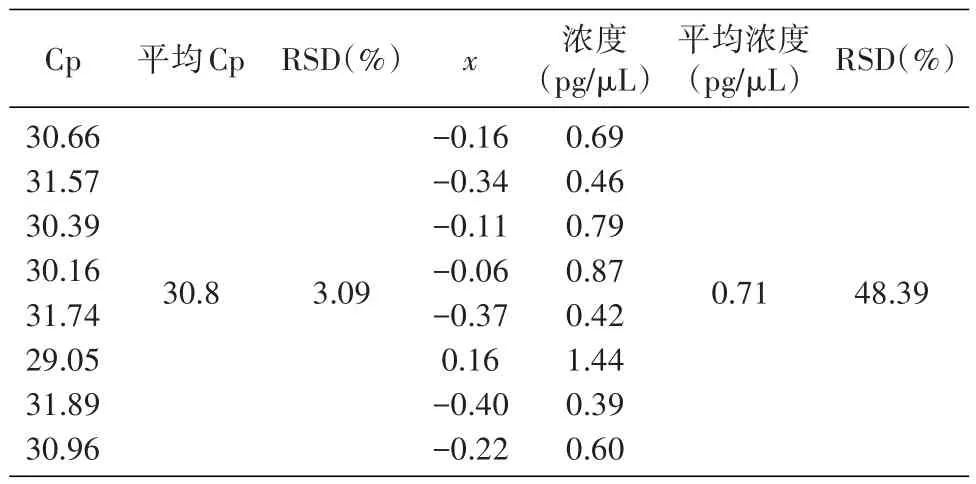
表1 用标准品测定精密度
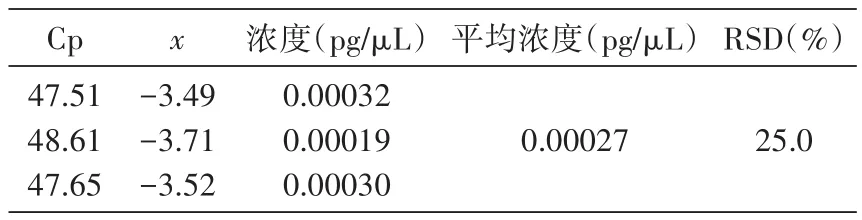
表2 用供试样品测定精密度
5批注射用重组新蛭素(酵母)外源DNA残留量测定结果见表5。根据真核细胞表达产品对宿主DNA残留量的标准,每剂量蛋白质产品中DNA残留量应小于100 pg。注射用重组新蛭素的包装规格为20 mg/支,使用方法是用1 mL注射用水溶解后静脉滴注。因此,注射用重组新蛭素中毕赤酵母DNA残留量应小于0.1 pg/μL。依据加样回收率测定结果,当DNA含量约为0.1 pg/μL时回收率为50.0%,因此,为增加样品测定结果的准确性,测定结果用标准品回收率校正计算样品DNA的残留量。5批重组新蛭素(酵母)外源DNA残留量分别为0.03、2.3、0.2、0.6和0.2 pg/mg,换算成每剂量依次为0.6、46、4.0、12、4.0 pg,均远小于每人份剂量100 pg的要求。
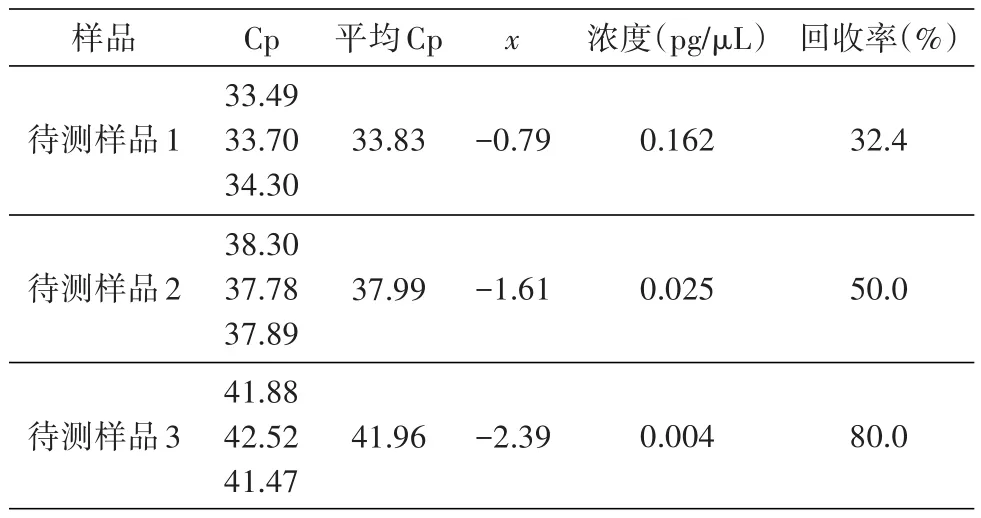
表3 加样回收率测定
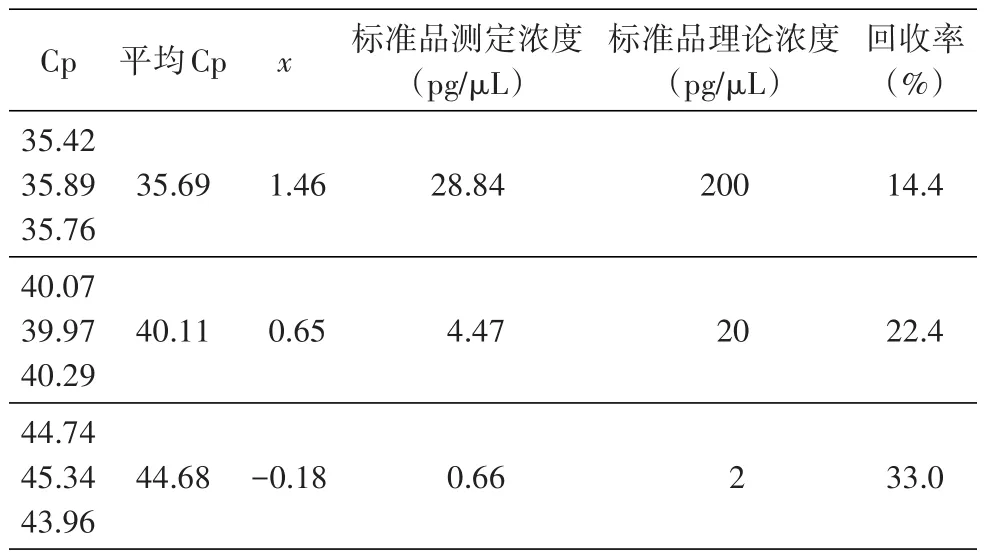
表4 标准品纯化后的回收率测定结果
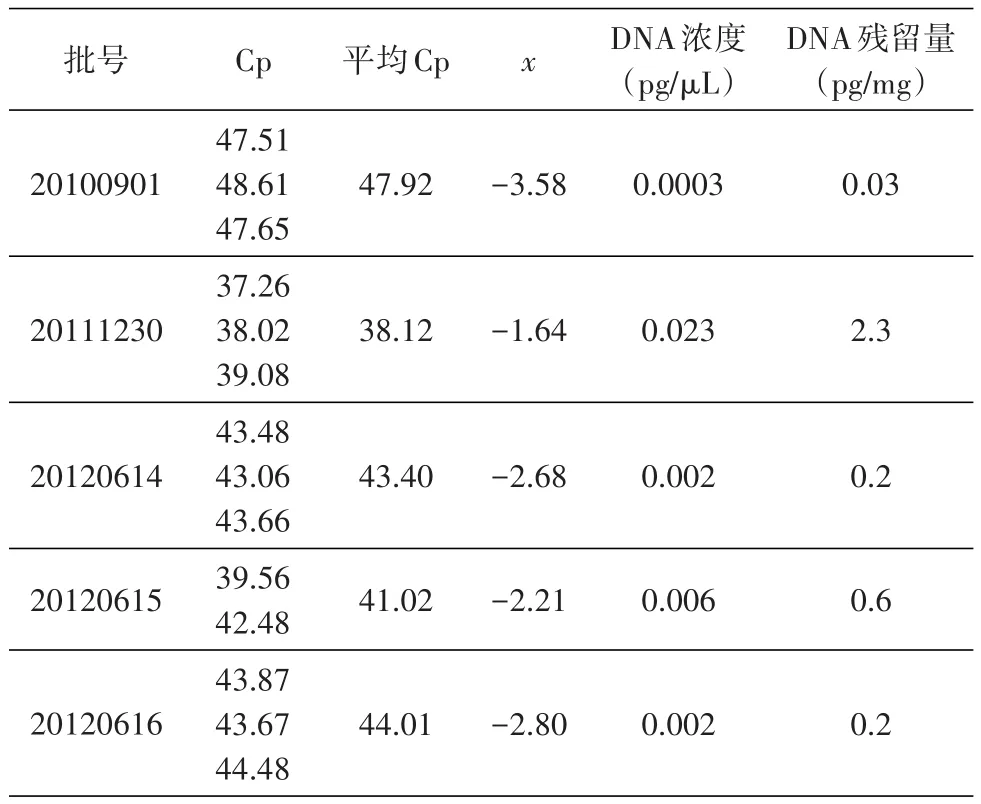
表5 注射用重组新蛭素DNA含量测定结果
3 讨论
与斑点杂交法相比,实时定量PCR法检测DNA具有操作简便、周期短、可定量分析等优点;与荧光法相比,具有更高的灵敏度,检出限可达1×10-1pg/μL水平。而且采用实时定量PCR法可在短时间内(30 min~2 h)完成对多个样本的检测,是高通量快速定量检测宿主基因组DNA残留的理想方法[16]。
毕赤酵母5S rRNA具有21个拷贝,分布于全部的4条染色体上,高拷贝数的特点使其很容易被检测到,而分布的广泛性使其即使在基因组不完整的情况下也能被检测到,因此,5S rRNA基因适宜作为酵母DNA残留检测的目标基因[15]。
实时定量PCR技术应用了荧光染料或探针来保证扩增的特异性,荧光信号的强弱同扩增产物的量成正比,从而实现了真正意义上的定量检测。SYBR GreenⅠ荧光染料无特异性,只要是双链就会结合,对PCR反应中的非特异性扩增或引物二聚体也会结合产生荧光,相对于TaqMan探针法,通常本底较高,精确度稍差。但SYBR GreenⅠ能够与所有双链DNA结合,因而对不同模板不需要定制不同的特异性探针,其通用性较好,而且价格相对较低,适合作为常规的质控方法。因此,在满足灵敏度要求的前提下,采取一定措施(如优化引物设计、优化PCR反应条件等)提高PCR反应的特异性,即可克服上述缺点,实现最佳的检测效果。
SYBR GreenⅠ荧光染料实时定量PCR技术检测毕赤酵母5S rRNA基因,当DNA含量为1×10-1~1000 pg/μL时呈现良好的线性关系,即使DNA含量低至1×10-4pg/μL也可以得到扩增,因此该方法具有较高的灵敏度,可用于微量DNA残留的检测。应用建立的检测方法对5批注射用重组新蛭素(酵母)产品中残留的宿主基因组DNA进行测定,5批产品的外源DNA残留量均符合小于100 pg/剂量的质量标准。由于DNA残留量均在检出限附近,因此RSD值相对较大。通过增加实验复孔数量和重复次数,能够得到比较稳定而准确的检测结果。该方法是一种灵敏度高、较准确、简便快速的方法,有较好的应用和推广前景。
[1]Staley C A,Huang A,Nattestad M,et al.Analysis of the 5'untranslated region(5'UTR)ofthe alcoholoxidase 1(AOX1)genein recombinantprotein expression in Pichia pastoris[J].Gene,2012,496(2):118-127.
[2]Chang S C,Hoang B,Thomas J T,et al.Cartilage-derived morphogenetic proteins new members of the transforming growth factor-β superfamily predominantly expressed in long bonesduringhuman embryonicdevelopment[J].BiolChem,1994,269(45):282-287.
[3]汪家政,范明.蛋白质技术手册[M].北京:科学技术出版社,2002:42-47.
[4]Wolter T R A.A ssays for controlling host-cell in purities in biopharmaceuticals[J].BioProcess Int,2005,3(2):40.
[5]国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社,2010:三部,252.
[6]国家药典委员会.中华人民共和国药典[M].北京:中国医药科技出版社,2010:三部,265,279.
[7]Kuroda S,Itoh Y,Miyazaki T,et al.A supersensitive dot-hy⁃bridization method rapid and quantitative detection of host-de⁃rived DNA in recombinant products at the one picogram level[J].Biochem Biophys Res Commun,1988,152(1):9.
[8]王军志.生物技术药物研究开发和质量控制[M].北京:科学出版社,2007:98.
[9]Lokteff M,Klinguer-Hamour C,Julien E,et al.Residual DNA quantification in clinical batches of BBG2Na,a recombi⁃nant subunit vaccine against human respiratory syncytial virus[J].Biologicals,2001,29(2):123.
[10]Briggs J,Panfili R.Quantitation of DNA and protein impuri⁃ties in biopharmaceuticals[J].Anal Chem,1991,63(9):850.
[11]Higuchi R,Fockler C,Dollinger G,et al.Kinetic PCR analy⁃sis:real-time monitoring of DNA amplification reactions[J].Nat Biotechnol,1993,11:1026-1030.
[12]Huong L V,Serge T,Huan H N,et al.A method for quanti⁃fication of absolute amounts of nucleic acids by(RT)-PCR and a new mathematical model for data analysis[J].Nucleic Acids Res,2000,28(7):18.
[13]Schmittgen T D.Real-time quantitative PCR[J].Methods,2001,25:383-385.
[14]张立国,张琚.实时定量PCR技术的介绍[J].生物技术,2003,13(2):39-40.
[15]De Schutter K,Lin Yao-Cheng,Tiels P.Genome sequence of the recombinantprotein production hostPichia pastoris[J].Nat Biotechnol,2009,27(6):561-569.
[16]Nissom P M.Specific detection of residual CHO host cell DNA by real-time PCR[J].Biologicals,2007,35(3):211.
猜你喜欢
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
广东茶业(2019年2期)2019-06-18 10:24:24
农药科学与管理(2019年12期)2019-05-20 09:33:26
中成药(2018年12期)2018-12-29 12:25:44
中成药(2018年1期)2018-02-02 07:20:31
中成药(2017年6期)2017-06-13 07:30:35
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
中国科技信息(2015年2期)2015-11-16 08:18:32
