
启动子Ptac与PsbA在鱼腥藻7120中表达hG-CSF的效率比较
2014-10-26 03:49:58宁文艳吴先敏王春梅陈伟东施定基
微生物学杂志 2014年3期
宁文艳,吴先敏,王春梅*,陈伟东,施定基
(1.北京中医药大学中药学院生物制药系,北京 100102;2.中国科学院植物研究所,北京 100093)
鱼腥藻7120(Anabaena sp.PCC7120)是一种丝状固氮蓝藻,是一类能进行光合放氧的原核生物[1],其作为模式植物被广泛用于光合、固氮、细胞分裂与分化等基础生命科学问题的研究。鱼腥藻7120基因组序列已于2001年测定完成[2]。由于蓝藻细胞可以利用简单无机物合成有机物,表达的外源基因产物不形成包涵体,并且多数蓝藻及其提取物对人畜无毒,因此有作为工业化生产基因工程药物宿主的潜力。目前,大批可用于蓝藻基因工程的载体已构建完成,一些外源基因也已在蓝藻细胞中成功表达,已成功表达的人源性因子有:人肿瘤坏死因子(tumor necrosis factor,TNF-α)、粒细胞集落刺激因子(granulocyte colony stimulating factor,G-CSF)、泛素融合的胸腺素α1(Ubiquitin biding Thymosinum α1,UB-α1)和人表皮生长因子(human epidermal grow factor,hEGF)等[3]。最近,蓝藻又成为合成生物学研究的热点,用于生产生物柴油等[4]。然而,蓝藻中外源基因的表达效率低,已成为其长足发展的瓶颈[5]。对于鱼腥藻7120中外源基因表达效率低的问题学者们从不同的角度进行了探索,转录水平启动子的调控是关键环节之一[6-7]。至今,已有内源性和外源性多种启动子 PsbA、PglnA、PrbcL、PrbcA、Ptac、PpetE、PpsbBⅠ、PpsbBⅡ及 PfurA 等被用于研究[8]。1993年ELHAI J证明启动子PsbA是鱼腥藻7120的内源强启动子,成为鱼腥藻7120穿梭表达载体最常用的启动子之一,如pRL488、pSBJ2和pKT-MT[9]。Tac启动子是大肠埃希菌中的强可诱导型启动子,用于蓝藻中表达外源基因也有较高的表达效率[10],虽然之前的研究表明光调节的PsbA和IPTG诱导的Ptac启动子是蓝藻中高效的启动子,但PsbA与Ptac两者在蓝藻中表达效率的比较研究未见报道。因此,本研究从转录水平和蛋白水平比较了2种启动子在蓝藻中表达外源基因hG-CSF的表达效率,为蓝藻基因工程的应用奠定基础。hG-CSF是全球销量最大的原核生物表达的基因工程药物,用于治疗化疗出现的白细胞减少症[11]。本研究选此基因作为研究对象,为利用蓝藻生产hG-CSF的研究提供参考。
1 材料与方法
1.1 材料
1.1.1 质粒与菌株 穿梭表达质粒pRL-489由美国密歇根大学Peter Wolk教授惠赠,穿梭表达质粒pRL-PsbA-GCSF和含Tac启动子表达的GCSF基因的穿梭表达质粒pRL-Tac-GCSF由北京中医药大学生物制药系实验室构建,大肠埃希菌DH5α、HB101和野生型鱼腥藻7120由北京中医药大学生物制药系实验室保存。
1.1.2 主要试剂与仪器 BamHⅠ、EcoRⅠ、HindⅢ等限制性内切酶和T4 DNA连接酶、PT-PCR试剂盒、ELISA试剂盒等购自TaKaRa公司;氨苄青霉素和卡那霉素抗生素、PCR扩增酶购自上海生工;质粒小量提取试剂盒及胶回收试剂盒购自北京博迈德生物技术有限公司;100 bp DNA Ladders购自北京全式金公司;琼脂糖试剂购于Promega公司;其他化学试剂均为国产分析纯。GeneAmp PCR System 9600(ABI公司);酶标检测仪(美国MD公司);DYY-6C型电泳仪(北京市六一仪器厂);HZQ-X100震荡培养箱(哈尔滨市东联电子技术开发有限公司)。
1.2 方法
1.2.1 蓝藻藻种培养 鱼腥藻7120生长于BG-11培养液中,藻种的纯化采用平板划线分离[12]。扩大培养时按1∶50的比例接种于50 mL三角瓶中,28 ℃、90 μmol·m-2·s-1光照条件下振荡培养[13]。
1.2.2 穿梭表达载体的构建 根据文献报道,设计并合成引物,P1:5'-CCGATAACGGTTCTGGCAAA-3'(HindⅢ酶切位点:AAGCTT),P2:5'-CGGGAAATTGTTATCCGCTCA-3'(BamHⅠ酶切位点:GGATCC)。采用分子克隆技术,以PGEX表达载体为模板,94℃预变性5 min;94 ℃变性30 s,68 ℃退火30 s,72 ℃延伸45 s,共30个循环;最后72℃延伸7 min,获得Tac启动子基因片段,产物经DNA胶回收试剂盒回收纯化,经HindⅢ与BamHⅠ双酶切,TA克隆PMD-19-T Simple Vector,送上海生物工程技术有限公司测序。针对pUC-GCSF载体上的GCSF基因设计引物:GCSF上游引物:5'-CGGGAGGAAGTCATATGACTCC-3'(酶切位点:BamHⅠGGATCC);GCSF下游引物:5'-CGTCATGGCTGGGCAAGG-3'(酶切位点:EcoRⅠ GAATTC)。测序正确的Tac启动子片段经HindⅢ与BamHⅠ双酶切后与载体pUC-G-CSF经双酶切进行大小片段连接,获得中间载体 pUC-tac-G-CSF,再经 HindⅢ、EcoRⅠ双酶切、Klenow Fragment补齐与 pRL489质粒经EcoRⅠ、BamHⅠ酶切,Klenow Fragment补齐后目的片段在T4连接酶的作用下进行平末端连接(由于2个载体间没有合适的确位点),最终得到穿梭表达载体pRL-tac-G-CSF。为确保所用样品具有相同量的RNA,rnpB(320 bp)基因,作为RT-PCR内参:rnpB上游引物:5'-ATAGTGCCACAGAAAAATACCG-3';rnpB下游引物:5'-AAGCCGGGTTCTGTTCTCTG-3'。
1.2.3 蓝藻的转化 采用三亲接合转移法[14]。①大肠埃希菌的准备:在三亲接合转移的前一天晚上,将含有穿梭表达载体的大肠埃希菌及含有辅助质粒pRL58和接合质粒RP4的大肠埃希菌接种到20 mL含有相应抗生素的LB培养液中,37℃培养过夜,4000 r/min离心5 min收菌,用普通LB培养液洗2次,重悬于5 mL普通LB培养液中,等体积混合2种菌液;②蓝藻的准备:取对数生长早期蓝藻培养物于4000 r/min离心2 min收集细胞,用培养液洗涤2次,再以适当体积的培养液悬浮藻细胞(使细胞终浓度约为108个/mL)。蓝藻细胞数的粗略计算公式:蓝藻细胞浓度(cells/mL)=OD665×4.5 ×107;③接合转移:将藻细胞与细菌按1∶10的比例混合均匀,轻轻地将其涂于无抗生素的固体培养基上,尽量使滤膜上的混合液均匀。正置平板1 h,使液体完全渗入培养基中,光照培养1~2 d,使蓝藻表达抗性,小心转移滤膜到含有相应抗生素(以50、100、150、200、250、300 mg/L的浓度梯度逐级递增卡那霉素的浓度)的平板上,继续光照培养。约2周后,出现阳性克隆。
1.2.4 转基因藻的分子检测 提取2种转基因鱼腥藻 pRL-PsbA-GCSF、pRL-Tac-GCSF质粒DNA,然后以质粒DNA为模板,用GCSF上、下游引物进行PCR扩增,凝胶电泳检测转基因藻中GCSF基因的存在。
1.2.5 转基因藻的RT-PCR 提取蓝藻总RNA,利用Trizol法分别提取野生型鱼腥藻7120,转基因型pRL-tac-GCSF(非诱导),转基因型pRL-tac-GCSF(1 mmol/L的IPTG诱导12 h),转基因型pRL-psbA-GCSF的总RNA,用DNAse除去基因组DNA后,分别用rnpB上、下游和G-CSF上、下游进行PCR。具体方法见PT-PCR使用说明书。
1.2.6 转基因藻蛋白的Elisa检测 取40 mL处于对数生长后期的野生型蓝藻PCC 7120、转基因型pRL-tac-GCSF(非诱导)、转基因型 pRL-tac-GCSF(1 mmol/L的IPTG诱导12 h)和转基因型pRL-psbA-GCSF藻液于离心管中,4000 r/min离心5 min收集藻细胞,弃上清,用裂解缓冲液洗涤2~3次,加入适量裂解液,-80℃、室温反复冻融6次,超声,12000 r/min离心5 min,收集上清,即为粗提蛋白。方法见人粒细胞集落刺激因子定量酶联检测试剂盒使用说明书。注意取等量(100 μg)总蛋白。
2 结果与分析
2.1 穿梭表达载体的构建
为了获得Ptac启动子,以PGEX表达载体为模板,进行PCR扩增,得到大小约100 bp的扩增产物。测序结果表明该片段长度为109 bp,与从文献中查阅的Tac序列比对,相似度为100%,说明序列完全正确。构建的穿梭表达载体pRL-tac-G-CSF(图1),转化大肠埃希菌DH5α,酶切鉴定,筛选阳性克隆。
2.2 转基因藻的获得
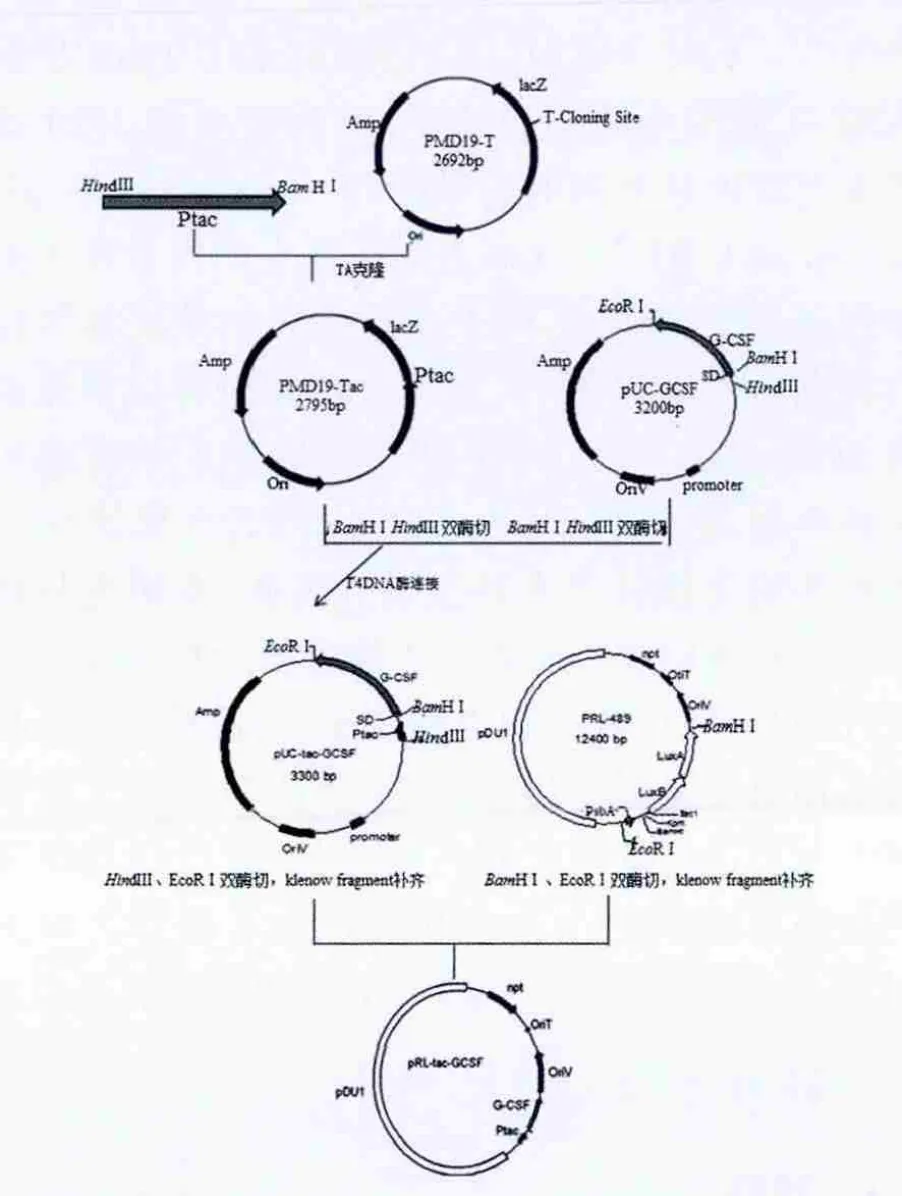
图1 穿梭表达载体pRL-tac-G-CSF的构建Fig.1 Scheme for construction of the shuttle expression vector pRL-tac-G-CSF
利用三亲接合转移[14],将穿梭表达载体pRL-489、pRL-PsbA-GCSF和pRL-tac-G-CSF分别转入鱼腥藻7120,在含卡那霉素的BG11固体平板上进行筛选。2周后,含质粒 pRL-489、pRL-PsbAGCSF和pRL-tac-G-CSF的鱼腥藻7120在膜上长出藻落(图2),而野生藻在抗生素压力下死亡。这一结果初步表明,质粒 pRL-489、pRL-PsbAGCSF和pRL-tac-G-CSF已分别转入鱼腥藻7120中,需进一步确认。
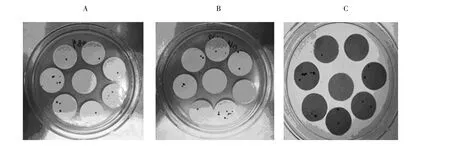
图2 转基因鱼腥藻7120的筛选Fig.2 Screening of the transgenic Anabaena sp.PCC 7120
2.3 转基因藻的分子检测
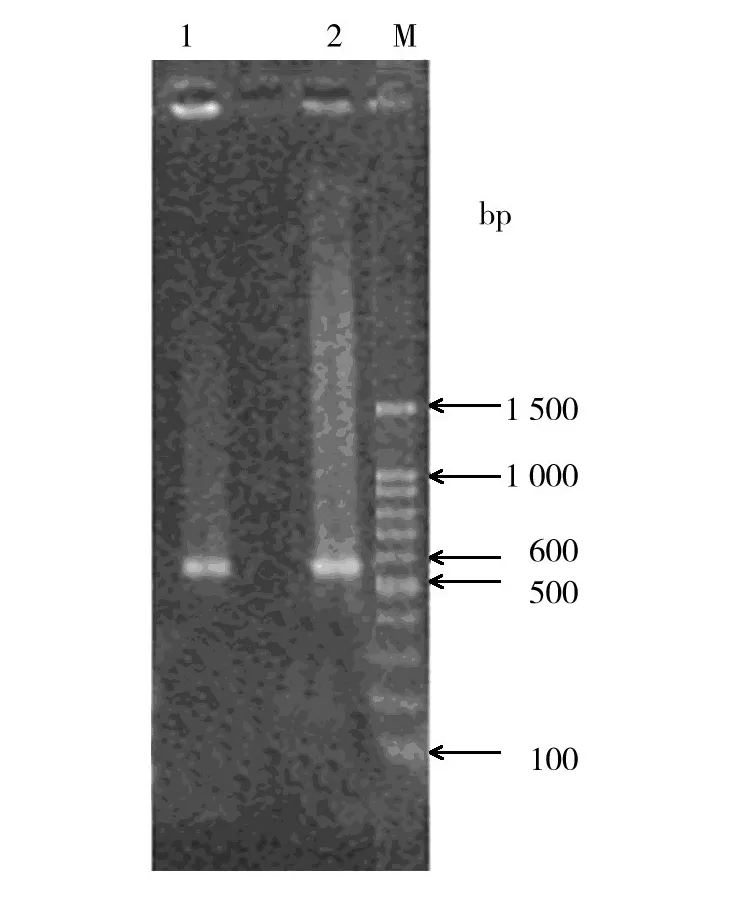
图3 转基因鱼腥藻7120的PCR检测Fig.3 Identification of PCR products in transgenic Anabaena sp.PCC 7120
提取2种转基因鱼腥藻 pRL-PsbA-GCSF、pRL-Tac-GCSF质粒DNA,然后以质粒DNA为模板进行PCR扩增,扩增结果显示,2种转基因鱼腥藻的质粒DNA能扩增出500 bp以上的G-CSF基因(图3),可初步断定基因转化成功。为了排除PCR非特异扩增的干扰,将转基因型蓝藻扩增出的条带纯化后,以hG-CSF上游引物作为测序引物,送上海生工测序。将测序结果与hG-CSF基因BLAST,证实确实为hG-CSF基因,因此,获得2种启动子驱动的hG-CSF转基因藻。
2.4 转基因藻的RT-PCR
为了检测转基因蓝藻中hG-CSF的转录水平,使用一步半定量RT-PCR方法检测mRNA转录水平。提取蓝藻总RNA,经1%琼脂糖凝胶电泳,结果可见,所提RNA条带清楚,但含有部分基因组DNA(图4A),会对后续的RT-PCR造成干扰;用DNAse除去基因组DNA后,经1%琼脂糖凝胶电泳,可见DNA除尽 (图4B),可用于RT-PCR。用 Random 6mers引物逆转录后,用rnpB基因作为内参,确保所用样品具有相同量的RNA,rnpB上、下游引物PCR后电泳,如图4C,条带亮度一致,可确定模板RNA用量一致。用G-CSF上、下游引物进行PCR,电泳,如图4D。结果以内参条带为参考经Qality ONE软件分析,并经统计学分析表明2种含hG-CSF的转基因藻均能转录 hG-CSF,但二者的转录水平差异不显著。
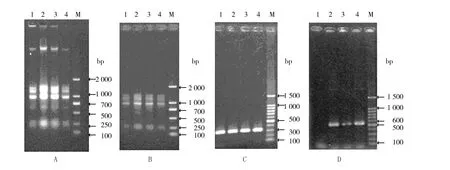
图4 转基因藻的RT-PCR结果Fig.4 The hG-CSF transcription level in transgenic Anabaena sp.7120 measured by RT-PCR method
2.5 转基因藻蛋白的ELISA检测

图5 转基因藻蛋白的ELISA分析Fig.5 Elisa analysis of the amount of hG-CSF protein in transgenic Anabaena sp.PCC 7120
为了进一步比较2种启动子在蓝藻中的最终作用效果,使用酶联免疫方法测定目的蛋白的表达量,结果见图5。由图5可知,7120-Tac-CSF在不诱导的条件下(Tac-),其表达量与野生型(7120)没有区别,即可认为是没有hG-CSF表达,诱导条件下表达量有上升趋势,7120-PsbA-CSF比Tac启动子在诱导条件下驱动的基因表达量高1.17 倍。
3 讨论
1980年以来,蓝藻基因工程快速发展,但其应用一直局限于理论研究,表达效率低是其发展瓶颈之一。近几年,蓝藻作为最理想的合成生物学底盘细胞之一引起了人们对蓝藻基因工程的广泛关注,但由于对蓝藻表达载体研究较少,缺少像大肠埃希菌那样丰富的表达载体成为其发展的局限因素[15-16],因此,对蓝藻基因表达元件及系统的研究尤其重要。
自1984年适用于接合转移的可在大肠埃希菌与蓝藻间穿梭表达的pRL载体构建完成[7]至今,杀蚊幼虫毒素蛋白、人的超氧化歧化酶、肿瘤坏死因子等大批外源基因已在蓝藻中成功表达[3]。然而,蓝藻基因工程产业化进程并不顺利,究其原因,外源重组蛋白表达效率低下可能是关键因素之一。近20年人们在转录、翻译及培养等方面进行了大量的研究工作,特别是对启动子的研究,推动了蓝藻基因工程的进展[17]。对于用于蓝藻中表达外源基因的启动子的研究,内源性的启动子有:光调节的 PsbA、cpcBA,温度调节cpcB1A1、PRPL,外源性的启动子有 IPTG诱导的Ptac、Ptrc,T7聚合酶识别的T7启动子,其中表达效率较高的启动子认为是IPTG诱导的Ptac和PsbA,本研究在最优IPTG诱导条件的结果说明PsbA的表达效率稍高于Ptac。由于利用Ptac可以比较容易通过添加诱导剂调整基因的表达水平,因此用于研究基因功能有较强的优势,本研究结果也表明,不加诱导剂的情况下,检测不到hGCSF的表达量,说明组成型表达极低。如果考虑工业应用,由于不需添加任何外源化学物质,蓝藻生长本身需要光照,光诱导的PpsbA启动子是理想的选择。
[1]Kushige H,Kugenuma H,Matsuoka M,et al.Genome-wide and heterocyst-specific circadian gene expression in the filamentous cyanobacterium Anabaena sp.strain PCC 7120[J].Journal of bacteriology,2013,195(6):1276-1284
[2]Kaneko T,Nakamura Y,Wolk C P,et al.Complete genomic sequence of the filamentous nitrogen-fixing cyanobacterium Anabaena sp.strain PCC 7120[J].DNA research,2001,8(5):205-213.
[3]席超,王春梅,施定基.蓝藻基因工程应用研究进展[J].中国生物工程杂志,2010,30(3):105-111.
[4]Wijffels R H,Kruse O,Hellingwerf K J.Potential of industrial biotechnology with cyanobacteria and eukaryotic microalgae[J].Current opinion in biotechnology,2013,24(3):405-413.
[5]魏兰珍.外源基因在集胞藻6803中高效表达及调控的研究[D].华东师范大学,2010.
[6]Chungjatupornchai W,Fa-aroonsawat S.The rrnA promoter as a tool for the improved expression of heterologous genes in cyanobacteria[J].Microbiological research,2014,169(5):361-368.
[7]陈伟东,王春梅,施定基.鱼腥藻7120遗传转化的研究进展[J].微生物学通报,2010,37(3):419-425.
[8]Huang H H,Lindblad P.Wide-dynamic-range promoters engineered for cyanobacteria[J].J.Biol.Eng,2013,22、7(1):10.
[9]Ma WM,Shi DJ,Wang QX,et al.Exogenous expression of the wheat chloroplastic fructose-1,6-bisphosphatase gene enhances photosynthesis in the transgenic cyanobacterium,Anabaena PCC7120[J].Journal of Applied Phycology,2005,17(3):273-280.
[10]Elhai J.Strong and regulated promoters in the cyanobacterium Anabaena PCC 7120[J].FEMS microbiology letters,1993,114(2):179-184.
[11]莫红楠,石远凯,孙燕.重组人粒细胞集落刺激因子在肿瘤化疗中应用20年回顾[J].中国新药杂志,2013,22(17):2027-2032.
[12]Ferris M J,Hirsch C F.Method for isolation and purification of cyanobacteria[J].Applied and environmental microbiology,1991,57(5):1448-1452.
[13]Castenholz R W.Culturing methods for cyanobacteria[J].Methods in enzymology,1988,167:68-93.
[14]Elhai J,Wolk C P.Conjugal transfer of DNA to cyanobacteria[J].Methods in enzymology,1988,167:747-754.
[15]杨键,游志庆,麦志茂,等.大肠杆菌-枯草杆菌穿梭载体的构建及其在蛋白酶基因表达中的应用[J].生物技术通报,2013(9):146-150.
[16]肖洁,郭刚,邹全明.提高大肠杆菌分泌表达重组蛋白的研究进展[J].微生物学杂志,2007,27(2):73-77.
[17]Zhou J,Zhang H,Meng H,et al.Discovery of a super-strong promoter enables efficient production of heterologous proteins in cyanobacteria[J].Sci.Rep.2014,4:4500.
猜你喜欢
湖泊科学(2022年4期)2023-01-04 14:20:54
东坡赤壁诗词(2022年2期)2022-04-15 02:32:43
东坡赤壁诗词(2019年5期)2019-11-14 10:36:10
当代水产(2019年9期)2019-10-08 08:02:42
当代水产(2018年8期)2018-11-02 05:30:42
上海农业学报(2016年5期)2016-02-10 06:53:01
中华灾害救援医学(2015年7期)2016-01-07 05:45:21
幼儿智力世界(2015年5期)2015-08-20 09:41:39
现代检验医学杂志(2015年2期)2015-02-06 02:00:56
现代检验医学杂志(2014年1期)2014-02-06 01:29:37
