
N-取代苯基四氢喹喔啉类化合物的合成工艺研究
2014-10-23 12:38陈莎娜周有骏
武汉轻工大学学报 2014年3期
陈莎娜,陈 新,周有骏,周 浩
(1.武汉轻工大学 生物与制药工程学院 湖北武汉 430023,2.中国人民解放军第二军医大学药学院,上海 200433)
自1947年多种苯取代 1,2,3,4-四氢喹喔啉类化合物用于抗疟疾活性研究以来[1],该类化合物引起了广泛关注。大量研究表明,1,2-二氢喹喔啉和1,2,3,4-四氢喹喔啉类化合物具有广泛的生理和药理活性,广泛应用于医药、化工等领域。如由于与四氢叶酸结构类似,可作为四氢叶酸类似物参与其功能调节[2];该类化合物能选择性抑制多种受体,调节多种生理病理活动,如可抑制细胞分裂素B1受体,缓解炎症和败血症引起的疼痛[3-4];抑制抗利尿激素 V2受体,增强利尿活性[5];可拮抗前列腺素D2受体,抑制过敏性炎症反应[6]等;作为一种有效的糖蛋白抑制剂,该类化合物可抑制 DC-SIGN[7],阻滞HIV、肝炎C等病毒的传播。此外,其类似物用于染料、有机半导体元件材料和细胞黏附剂等的研究和应用报道也屡见不鲜。鉴于该类化合物是应用于多个领域的重要化工中间体,因此,对于该类化合物的合成方法研究也受到广泛重视。
目前,该类化合物的合成方法已有诸多报道(scheme1)。其中,以邻苯二胺与β-二羰基化合物,酮[8]、α-卤代酮[9]、α-羟基取代酮[10],环氧化合物[11]等在强碱性或酸性条件下缩合形成喹喔啉杂环,再进行还原是最为常见的合成方法。但目前对于该类N取代化合物的制备通常副产物较多,收率较低,对N取代反应缺乏选择性。此外也有研究者以邻苯二胺环合成 2-氧代 1,2-二氢喹喔啉环[12],再进行还原得到2-芳基取代四氢喹喔啉化合物。虽然此方法能对N取代反应具有一定的选择性,但仍不理想,且起始原料价格较昂贵,并需要LiAlH4等强还原剂才能顺利进行。因此,笔者设计了一条以邻苯二胺和β-溴代苯乙酮为起始原料,经环合、N取代及还原三步反应选择性制备N-取代苯基四氢喹喔啉类化合物的新合成路线,并进行合成工艺优化研究。
1 仪器和试剂
集热式恒温磁力搅拌器(上海梅颖普仪器有限公司);旋转蒸发仪(上海予华仪器设备有限公司);循环水式多用真空泵(上海诚献仪器设备有限公司);ZF7三用紫外分析仪(巩义市予华仪器设备有限责任公司);Burker ACF-300(TMS为内标)核磁共振仪。所用试剂均为市售分析纯。
2 合成路线设计
合成路线如下。

3 实验步骤
3.1 2-苯基-1,2-二氢喹喔啉(a)的合成[13]
在 N2保护下,加邻苯二胺 1.1 g(10.19 mmol),β-溴代苯乙酮 2 g(10.01 mmol),碳酸钠 0.83 g(10.12 mmol)和甲醇20 mL于100 mL圆底烧瓶中,室温搅拌18 h,TCL检测反应完全。反应液过滤,滤饼用少量水洗,干燥,得黄色固体(1.92 g,91.43%)。1H NMR(300 MHz,DMSO) δ 7.94(s,2H),7.49(s,3H),7.12(d,J=6.9 Hz,1H),6.92(d,J=6.6 Hz,1H),6.58(d,J=7.4 Hz,2H),6.23(s,1H),4.37(s,2H).MS-ESI(m/z):209(M+1)。
3.2 N-肉桂酰-3-苯基-1,2-二氢喹喔啉(b)的合成[14]
加1 g(4.8 mmol)a,碳酸钾溶液5 mL(1.8 M,9 mmol)于15 mL丙酮中,冰浴搅拌下,缓慢分批加入肉桂酰氯1.04 g(6.27 mmol),待反应液全部由橙色变为黄色,TCL检测示反应完全,反应终止。将反应液过滤,加少量水润洗,得黄色固体 b(1.37g,84.31%)。1H NMR(300 MHz,DMSO) δ 8.10(d,J=3.9 Hz,2H),7.70(s,1H),7.67 – 7.59(m,2H),7.59 – 7.47(m,4H),7.40(d,J=3.2 Hz,4H),7.36 – 7.28(m,2H),7.07(d,J=15.0 Hz,1H),4.95(s,2H)。MS-ESI(m/z):339(M+1)。
3.3 N-肉桂酰-3-苯基-1,2,3,4-四氢喹喔啉(c)的合成
加0.5 g(1.5 mmol)b溶解于5 mL四氢呋喃和5 mL乙醇中,加适量硼氢化钠,于60℃下回流,TCL监测示反应完全。反应液加水淬灭,减压浓缩除去大部分溶剂,水溶液用乙酸乙酯萃取3次,合并有机相,加无水硫酸钠干燥,过滤,滤液减压浓缩至干。将棕黄色固体进行硅胶柱层析(石油醚/乙酸乙酯=10∶1),得黄色固体 c(0.43 g,87.75%)。1H NMR(300 MHz,DMSO) δ 7.50(s,2H),7.47 – 7.34(m,J=5.1 Hz,4H),7.34 – 7.25(m,4H),7.25 – 7.17(m,J=4.8 Hz,1H),7.09 – 6.93(m,2H),6.91(s,1H),6.80(d,J=8.0 Hz,2H),6.56(t,J=7.6 Hz,1H),4.67(s,1H),3.91(s,2H).MS-ESI(m/z):341(M+1)。
4 正交试验设计
在2-苯基-1,2-二氢喹喔啉(a)的合成过程中,由于该中间体不稳定,容易脱氢形成稳定的喹喔啉芳香化合物a'(如图1)。按照文献[3]报道的方法,以乙酸钠为催化剂,在CO2气体保护下加热回流,主产物并不是目标化合物a,而是芳香化的喹喔啉化合物a'。因此,我们针对反应涉及到的催化剂,温度和溶剂三要素,进行了实验条件优化。
首先尝试分别以乙酸钠和碳酸钠为催化剂,降低反应温度,考察反应情况,结果发现以碳酸钠为催化剂,降低温度不仅收率得到大大提高,而且简化了后处理过程(见表1)。

表1 催化剂对反应收率的影响
据此,笔者采用碳酸钠为催化剂,针对反应涉及到的反应温度(A)和溶剂(B)两要素,进行了两因素三水平的正交试验设计,按照L9(34)正交表安排9个反应,考察此二因素对目标产物a收率的影响,以进一步优化实验方案。研究因素水平和实验安排见表2和表3。

图1 2-苯基-1,2-二氢喹喔啉(a)合成的可能副产物
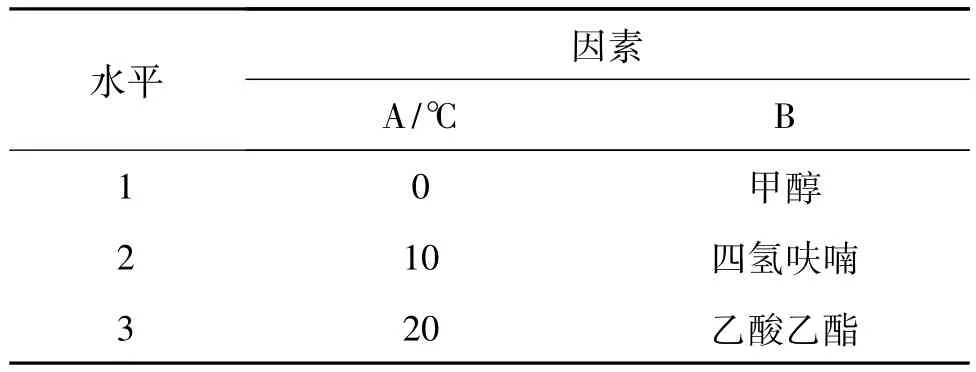
表2 制备2-苯基-1,2-二氢喹喔啉的影响因素水平表

表3 制备2-苯基-1,2-二氢喹喔啉的正交试验结果
5 结果与讨论
5.1 2-苯基-1,2-二氢喹喔啉(a)的合成
在分别以乙酸钠和碳酸钠做催化剂,甲醇为溶剂进行室温反应时,收率分别为 79.52%和91.43%,证实以碳酸钠作为催化更有利于目标产物a的稳定存在,杂质更少,收率更高。推测可能是乙酸钠在甲醇中的溶解度大于碳酸钠所致。以碳酸钠进行正交实验,实验结果(见表3)和正交实验方差分析(见表4)表明:两个因素均影响目标化合物a的收率,且A>B,最佳实验条件为A1B1。分析实验结果显示,(1)低温有利于产物的吸出和稳定存在。在室温下,反应能快速进行,副产物a'的浓度较小;提高反应温度,反应液颜色加深,副产物a'浓度增大,而降低反应温度,尽管反应收率很高,但反应时间需延长到40 h。(2)实验结果发现,随着反应溶剂极性不断降低,尽管反应转化率没有很大变化,但收率却大大下降。原因在于随着目标产物溶解度的增大,大量产物溶解在溶液中而不能析出,而需要进一步纯化。因此,综合考虑以甲醇为溶剂,在10℃下反应18 h,不仅反应时间合适,降低了时间成本,而且收率较高,为最优实验方案。

表4 化合物a制备的正交试验方差分析
5.2 N-肉桂酰-2-苯基-1,2-二氢喹喔啉(b)的合成
在反应后处理时,按照文献[14]报道的方法,将反应液倾入冰水中,有黏稠状物质形成,而没有固体析出,分离纯化困难,因此我们尝试加少量溶剂,使反应产物直接析出。另外,缓慢分批加入肉桂酰氯,更有利于反应能快速进行。综上所述,笔者设计了一条以邻苯二胺和β-溴代苯乙酮为起始原料,经环合、N取代及还原三步反应选择性制备N-取代苯基四氢喹喔啉类化合物的新合成路线,并对关键中间体2-苯基-1,2-二氢喹喔啉的环合反应进行了平行实验和正交实验设计研究。结果表明,以碳酸钠为催化剂,甲醇为溶剂进行室温反应,为环合反应的最优实验方案,收率达91.43%。新合成路线的总收率达67.64%。新合成方法步骤简单,条件温和,收率较高,适用于大量制备。
[1]Cavagnol J C.,Wiselogle F Y.1-Alkyl-1,2,3,4-tetrahydroquinoxalines[J].J Am Chem Soc,1947,69:795-799.
[2]Benkovic S J,Benkovic P A,Comfort D R.Models for tetrahydrofolic acid.I.Condensation of formaldehyde with tetrahydroquinoxaline analogs[J].J Am Chem Soc,1969,91,5270-5279.
[3]Morissette G.,Fortin J P,Otis S,et al.A novel nonpeptide antagonist of the kinin B1receptor:Effects at the rabbit receptor[J].J Pharmacol Exp Ther,2004,311:1121-1130.
[4]Chen J J,Qian W,Biswas K,et al.Discovery of dihydroquinoxalinone acetamides containing bicyclic amines as potent Bradykinin B1 receptor antagonists[J].Bioorg Med Chem Lett,2008,18,4477-4481.
[5]Gluchowski C.(2-Imidazolin-2-ylamino)tetrahydroquinoxalines and methods for using same[P].EP:0422878 A1,1991-04-17.
[6]Torisu K,Kobayashi K,Iwahashi M,et al.Discovery of a new class of potent,selective and orally active prostaglandin D2 receptor antagonists[J].Bioorg Med Chem,2004,12:5361 –5378.
[7]BorrokM J,Kiessling L L..Non-carbohydrate inhibitors of the lectin DC-SIGN[J].J Am Chem Soc,2007,129,12780-12785.
[8]Cho C S,Ren W X,Shim S.C.Ketones as a new synthon for quinoxaline synthesis[J].Tetrahedron Lett,2007,48:4665-4667.
[9]Das B,VenkateswarluK,Sunee K,et al.An efficient and convenient protocol for the synthesisof quinoxalines and dihydropyrazines via cyclization-oxidationprocesses using HClO4-SiO2as a heterogeneousrecyclable catalyst[J].Tetrahedron Lett 2007,48:5371-5374.
[10]Pan Fan ,Chen Tang-ming ,Jia-jia,et al.Ga(ClO4)3-catalyzed synthesis of quinoxalines by cycloadditionof a-hydroxyketones and o-phenylenediamines[J].Tetrahedron Lett,2012,53:2508-2510.
[11]Antoniotti S,Dunachb E.Direct and catalytic synthesis of quinoxaline derivatives from epoxides and ene-1,2-diamines[J].Tetrahedron Lett,2002(43):3971-3973.
[12]Smith R F,Rebel W J,Beach T S.Convenient syntheses of 1,2,3,4-tetrahydroquinoxalines[J].J Org Chem,1959,24(2):205-207.
[13]Kolos N N.,Insuasti B,Kiroga K,et al.3-Aryl-1,2-Dihydroquioxalines[J].Chemistry of Heterocyclic Compounds,1986:918-922;
[14]Mickelson J W,Jacobsen E J,Carter D B,et al. High-Affinity α-Aminobutyric Acid A/Benzodiazepine Ligands: Synthesis and-Structure-Activity Relationship Studies of a New Series of TetracyclicImidazoquinoxalines[J].J Med Chem,1996,39,4654-4666.
猜你喜欢
妇女生活(2020年4期)2020-05-19
农药科学与管理(2019年8期)2019-11-23
化工环保(2017年5期)2017-10-31
中学化学(2017年2期)2017-04-01
新乡学院学报(2016年6期)2016-12-01
红外技术(2016年9期)2016-03-27
化学教学(2015年8期)2015-10-15
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
中国氯碱(2014年10期)2014-02-28
江西理工大学学报(2013年1期)2013-03-20
