
梓醇对缺糖缺氧诱导PC12细胞损伤的保护作用研究
2014-10-10 02:14:12王莹刘玉刚陈占法
河北医药 2014年1期
王莹 刘玉刚 陈占法
脑组织代谢旺盛,血流量丰富,因此脑缺血极易引起神经细胞损害,致功能缺失。缺血、缺氧条件下,神经元产生大量自由基,同时钙离子(Ca2+)超载,损伤线粒体,从而诱导神经元凋亡[1]。梓醇是一种环烯醚菇类的小分子化合物。研究表明,能促进PC12细胞轴突生长、保护海马神经元等[2,3]。文献报道连二亚硫酸钠联合无糖Earle’s液损伤模型能较好地反映神经细胞缺糖缺氧损伤[4],本研究以PC12细胞为基础建立该模型,考察梓醇的干预作用,并从拮抗自由基损伤和减轻Ca2+超载探讨其作用机制。
1 材料与方法
1.1 材料 大鼠肾上腺嗜铬细胞瘤细胞(PC12)购自中科院上海细胞库;梓醇购自上海同田生物技术公司,纯度≥97%;连二亚硫酸钠、RPMI1640培养基、噻唑蓝MTT、胎牛血清均为 Gibco公司产品;乳酸脱氢酶(LDH)试剂盒、超氧化物歧化酶(SOD)试剂盒、谷胱甘肽过氧化物酶(GSH-Px)试剂盒、丙二醛(MDA)试剂盒购自南京建成生物公司。
1.2 方法
1.2.1 PC12细胞培养及分组:PC12细胞培养于含10%胎牛血清的RPMI1640培养基。细胞经0.05%胰蛋白酶消化传代后置含5%CO2的37℃培养箱内培养,取对数生长期的细胞进行实验。实验共分5组:正常对照组:不加任何药物;缺糖、缺氧损伤组:加入终浓度含0.5 mmol/L连二亚硫酸钠的无糖Earle’s液;梓醇高、中、低剂量组:梓醇终浓度分别为 10、1、0.1μmol/L,各浓度梓醇预处理细胞24 h后,加入连二亚硫酸钠损伤,1 h后进行指标检测。
1.2.2 MTT法检测细胞存活率:细胞以1×105个/ml接种于96孔板,每孔100μl。12 h后,按照实验分组给予处理。检测时每孔加5 mg/ml MTT溶液15μl,继续培养4 h,弃上清后每孔加入150μl DMSO,振荡10 min,用酶标仪在492 nm波长下测定各孔吸光度值(OD值)。细胞存活率(%)=处理组OD值/对照组OD值×100%。实验重复3次。
1.2.3 乳酸脱氢酶(LDH)漏出率检测:细胞以4×105个/ml接种于24孔板,每孔500μl。12 h后,按照实验分组给予处理。检测时,按照试剂盒说明书进行操作。LDH释放率(%)=上清LDH活性/(上清LDH活性+细胞溶解液LDH活性)×100%。
1.2.4 SOD、GSH-Px活性及MDA含量测定:细胞以4×105个/ml接种于24孔板,每孔500μl。12 h后,按照实验分组给予处理。检测时,收集细胞,超声裂解后低温离心,取上清液按照试剂盒说明书测定SOD、GSH-Px活性及MDA含量。
1.2.5 细胞内游离 Ca2+浓度测定[5,6]:细胞以 7 ×104个/ml接种于25 cm2细胞培养瓶中,每瓶5 ml。12 h后,按照实验分组给予处理。检测时,收集细胞,应用基础培养基进行漂洗,加入1μmol/L Fura-2/AM,37℃避光恒温震荡45 min,漂洗,于双波长荧光分光光度计上340 nm和380 nm处测F值,分别加入Triton X-100和EGTA,测F最大值(Fmax)及最小值(Fmin)。Ca2+=Kd(R-Rmin)/(Rmax-R)(Fmin/Fmax)。其中Kd是Fura-2与Ca2+的解离常数,为224 nmol/L;R=F340/F380。
1.3 统计学分析 应用SPSS 16.0统计软件,计量资料以±s表示,组间比较采用单因素方差分析检验,P<0.05为差异有统计学意义。
2 结果
2.1 PC12细胞生存率 与正常对照组比较,模型组细胞生存率显著降低(P<0.01);与模型组比较,梓醇1μmol/L组、10μmol/L组细胞生存率显著升高,差异有统计学意义(P<0.01)。见表1。
表1 梓醇对PC12细胞生存率影响n=9,%,±s

表1 梓醇对PC12细胞生存率影响n=9,%,±s
注:与正常对照组比较,*P<0.01;与模型组比较,#P<0.01
生存率正常对照组组别100模型组 46.8±1.9*梓醇0.1μmol/L组 49.1±3.6梓醇1μmol/L组 60.8±1.5#梓醇10μmol/L组 71.0±1.5#
2.2 LDH漏出率 与正常对照组比较,模型组LDH漏出率显著升高(P<0.01);与模型组比较,梓醇可剂量依赖性的降低LDH漏出率(P<0.05或<0.01)。见表2。
2.3 SOD、GSH-Px的活性及MDA含量 与正常对照组比较,缺糖缺氧损伤降低了细胞上清SOD、GSH-Px的活性(P <0.01),而MDA含量显著升高(P <0.01)。与模型组比较,梓醇1μmol/L组、10μmol/L组可剂量依赖性的升高SOD活性(P<0.01),升高GSH-Px活性(P <0.05或<0.01),梓醇10μmol/L组可降低MDA含量(P <0.05)。见表3。

表2 梓醇对PC12细胞LDH漏出率影响
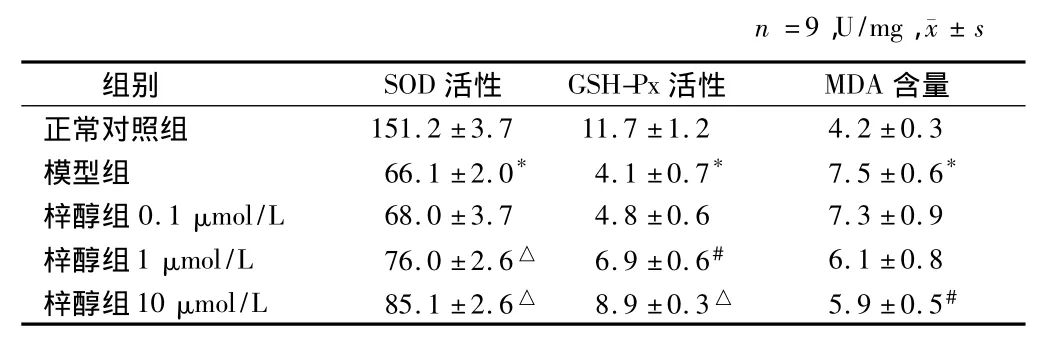
表3 梓醇对PC12细胞SOD、GSH-Px活性及MDA含量影响
2.4 细胞内游离Ca2+浓度 与正常对照组相比,模型组细胞内游离Ca2+浓度显著增加(P<0.01)。与模型组比较,梓醇可剂量依赖性的降低细胞内游离Ca2+浓度(P <0.05或 <0.01)。见表4。

表4 梓醇对PC12细胞内游离钙离子浓度影响
3 讨论
PC12细胞为大鼠肾上腺髓质嗜铬细胞瘤克隆化的细胞株,具有较典型的神经内分泌细胞特征,已广泛用于细胞信号传导与细胞内信使、兴奋性神经毒性和神经保护机制的研究[7,8]。
缺血产生的脑细胞损伤在本质上主要是脑组织氧、糖缺乏而引发的一系列事件的结果。体外常用缺氧低糖培养引起神经细胞损伤模拟脑缺血模型[9]。实验中主要选择性的去除具有至关重要作用的葡萄糖以代表缺血所致的低营养条件。加入连二亚硫酸钠清除培养基质中的氧而达到使细胞发生化学性缺氧的目的,研究表明,含0.5 mmol/L连二亚硫酸钠的无糖Earle’s液与PC12细胞共培养1 h,细胞活力下降。梓醇预给药24 h,能减轻缺糖、缺氧对 PC12细胞的损伤。
正常情况下LDH存在于细胞液中,一旦细胞膜通透性升高,即释放到细胞外,因此LDH释放是细胞膜通透性升高的一种标志性蛋白[10]。本实验结果显示,缺糖、缺氧导致PC12细胞膜通透性增加,LDH大量释放。梓醇可剂量依赖性的减少细胞LDH的漏出,表明梓醇对缺糖、缺氧致细胞损伤具有一定的保护作用。
正常代谢产生的自由基可被体内相应的自由基清除酶如SOD,GSH-Px及时清除,这对于保护细胞不受毒性氧自由基损伤有重要作用[11]。脑缺血再灌注时,产生大量氧自由基、激活兴奋性氨基酸NMDA受体、促使内源性谷氨酸大量释放而引发神经毒性,同时氧自由基防御系统功能受损,影响SOD、GSH-Px的活性,机体清除氧自由基的能力减弱,因此,测定SOD、GSHPx活性不仅可反映神经细胞损伤的原因,而且还可以判断细胞损伤的程度[12]。MDA是脂质过氧化反应中的重要产物,测定MDA生成可反映细胞膜结构脂质过氧化反应的程度,间接反映氧自由基生成的情况以及细胞损伤的程度[13]。因此,我们分别检测了SOD、GSH-Px及MDA在细胞中的活性及含量变化。
本实验表明,细胞损伤后,SOD、GSH-Px活性显著下降,MDA含量显著升高,表明缺血再灌注后氧自由基清除能力下降,氧自由基生成增加,脂质过氧化程度加剧,提示所致细胞损伤与氧化损伤有关。缺血前加入梓醇,可剂量依赖性提高细胞中SOD、GSH-Px活性,降低MDA含量。提示梓醇可降低细胞脂质过氧化反应,提高细胞清除超氧阴离子的能力,从而发挥抗自由基损伤的作用。
Ca2+是神经细胞信息传递的重要第二信使,对于维持神经细胞正常代谢与功能起着重要的调控作用。细胞缺氧时,神经元产生大量自由基,细胞线粒体功能障碍,能量供应不足,钙泵不能主动把细胞内的Ca2+泵出细胞外,由于ATP不足,糖酵解代偿性增强,使细胞内[Ca2+]i升高,导致细胞Ca2+超载,启动一系列病理生理机制,造成细胞损伤或死亡[14,15]。同时,Ca2+超载又会加重自由基的产生。体内实验已表明Ca2+在缺血缺氧性脑损伤中起着重要的作用[16]。本实验也显示,缺血缺氧神经细胞内Ca2+含量明显高于正常对照组,说明PC12细胞在缺血缺氧的情况下,细胞膜受到损伤,其通透性发生了改变,出现了Ca2+内流。梓醇可显著降低受损PC12细胞内的Ca2+含量,抑制Ca2+内流,维持胞内Ca2+稳态平衡;提示其有可能通过抑制细胞内Ca2+超载而对缺血缺氧损伤的PC12起到一定的保护作用。
综上所述,梓醇对连二亚硫酸钠诱导的缺糖缺氧细胞损伤具有保护作用,该作用可能与其清除自由基和抑制钙超载有关。
1 兰希发,姚文秀,郭阳.脑缺血再灌注后神经细胞凋亡的机制.中国临床康复,2003,7:2726-2727.
2 Li DQ,Li Y,Liu Y,et al.Catalpol prevents the loss of CA1 hippocampal neurons and reduces working errors in gerbils after ischemia-reperfusion injury.Toxicon,2005,46:845-851.
3 Jiang B,Liu JH,Bao YM,et al.Catalpol inhibits apoptosis in hydrogen peroxide-induced PC12 cells by preventing cytochrome release and inactivating of caspase cascade.Toxicon,2004,43:53-59.
4 刘娜,左萍萍,周帆,等.连二亚硫酸钠致PC12和NG108215细胞拟缺血损伤研究.中国药理学通报,1998,14:525-529.
5 Chen JG,Backus KH,Deitmer JW.Intracellular calcium transients and potassium current oscillations evoked by glutamate in cultured rat astrocytes.JNeurosci,1997,17:7278-7287.
6 Grynkiewicz G,Poenie M,Tsien RY.A new generation of Ca2+indicators with greatly improved fluorescence properties.J Biol Chem,1985,260:3440-3450.
7 Fukada K,Shoda T,Mima H,et al.Midazolam induces expression of c-Fos and EGR-1 by a non-GABA ergic mechanism.Anesth Analg,2002,95:373-378.
8 Shinomiya N,Shinomiya M.Dichlorodiphenyltrichoethane suppresses neurite outgrowth and induces apoptosis in PC12 pheochromocytoma cells.Toxicol letters,2003,137:175-183.
9 Otter D,Austin C.Hypoxia,metabolic inhibition,and isolated rat mesenteric tone:influence of arterial diameter.Microvascular Res,2000,59:107-114.
10 Lobner D.Comparison of the LDH and MTT assays for quantifying cell death:validity for neuronal apoptosis.J Neurosci Methods,2000,96:147-152.
11 Mugge A,Elwell J,Peterson TE,et al.Release of intact endothelium-derived relaxing factor depends on endothelial superoxide dismutase activity.Am JPhysiol,1991,260:219-255.
12 Rodrigoa J,Femaez AP,Semanoa J,et al.The role of free radicals in cerebral hypoxia and ischemia.Free Radic Biol Med,2005,39:26-50.
13 Kinnula VL,Everitt JI,Whorton AR,et al.Hydrogen peroxide productionby alverlar type Ⅱ cells,alveolar macrophages and endothelial cells.Am J Physiol,1999,261:84-91.
14 Weber JT.Calcium homeostasis following traumatic neuronal injury.Curr Neurovasc Res,2004,1:151-171.
15 Vannucci RC,Brucklacher RM,Vannucci SJ.Intracellular calcium accumulation during the evolution of hypoxicischemic brain damage in the immature rat.Brain Res Dev Brain Res,2001,126:117-120.
16 Sask IC,Kitagaw AH,Zhang WR,et al.Temporal profile of cytochrome Cand caspase-3 immunoreactives and TUNEL staining after permanent middle cerebral artery occlusion in rat.Neural Res,2000,22:223-228.
猜你喜欢
天津医药(2023年7期)2023-07-17 09:26:34
中国骨质疏松杂志(2022年6期)2022-07-05 09:34:36
中成药(2021年5期)2021-07-21 08:39:04
硫酸工业(2020年10期)2020-12-10 05:20:10
医药导报(2020年4期)2020-04-29 09:24:04
中西医结合心脑血管病杂志(2018年7期)2018-05-11 03:30:30
癌变·畸变·突变(2016年3期)2016-02-27 06:15:26
发明与创新(2015年37期)2015-02-27 10:40:25
食品工业科技(2014年15期)2014-03-11 18:17:24
癌变·畸变·突变(2014年3期)2014-03-01 04:39:48
