
联苯二酚尾饰锌卟啉的合成及其光谱性质研究*
2014-09-30 09:42江国防牛平平张志超焉保明郭灿城
湖南大学学报(自然科学版) 2014年1期
江国防,牛平平,张志超,焉保明,郭灿城
(湖南大学 化学化工学院,湖南 长沙 410082)
卟啉是具有很好生物活性的大分子,广泛存在于自然界中,如构成血红蛋白细胞色素的铁卟啉以及构成叶绿素的镁卟啉.它们是生物活性分子的核心部分,参与生物体内系列重要活动.卟啉分子具有刚柔性、电子缓冲性、光电磁性和高度的化学稳定性,将某些具有特殊性质的基团连在卟啉环上合成一些具有复合功能的卟啉化合物日益受到人们的重视[1-3].
卟啉化合物具有良好的光敏性,对癌细胞有独特的亲和力,因此在医学上可以用做癌症的检测和治疗.随着对卟啉的深入研究,以卟啉类的光敏剂为核心的光动力疗法已经产生.近些年,利用卟啉特有的性能和结构来进行功能分子的合成、设计和应用研究,受到了生物学、医学以及化学等领域的关注[4-6].
光动力治疗(photodynamictherapy,PDT)是一种疗效好、创伤小的肿瘤临床治疗方法.该疗法通过选择性结合病变组织、在光照下产生活性氧杀灭癌细胞而达到治疗目的.高效光敏剂的选取是决定光动力治疗效果的关键[7].卟啉类化合物具有特殊的生物学功能,对于发生异常增殖的肿瘤细胞具有独特的亲和力,同时卟啉能够特异地在肿瘤处发生富集.近年来,人们尝试将卟啉类化合物与抗癌类药物进行共价联接,从而制备出许多有靶向效应的抗肿瘤药物,且疗效不错.如:将卡铂(能抑制脱氧核糖核酸)与四苯基卟啉连接,产物不仅会在肿瘤处发生富集,而且提高了其穿透力,增加了其光敏剂的吸收波长,疗效明显增强[8-9].此外还有替加氟[10]、甲氨蝶呤[11]、以及氟尿嘧啶[12]与卟啉形成的的键联化合物.卟啉化合物之所以能够用作抗肿瘤药物,是因为其对肿瘤组织有独特的亲和力和较高的选择性[13-14].本文报道了由5-(4-羟基苯基)-10,15,20-三苯基卟啉出发经过四步反应合成了联苯二酚尾饰锌卟啉4,合成路线如图1所示.我们同时研究了所合成的目标化合物的光谱性能,结果表明该目标化合物是一类良好的光敏剂,在光动力治疗方面具有潜在的应用价值.

图1 联苯二酚尾饰锌卟啉合成路线Fig.1 Schemel synthetic route of zinec oprphyrins tailed by 4,4'-biphenyldiol
1 实验部分
1.1 试剂与仪器
INOVA-400型核磁共振仪(美国 Varian公司);Hitachi F-4500型荧光光谱仪(日立公司);UV-2450紫外光谱仪(日本岛津公司);所用试剂均为市售分析纯试剂,要求无水的溶剂均经去水和重蒸处理.5-对羟基苯基-10,15,20-三苯基卟啉参照文献[15]方法合成.
1.2 5-[4-(5-溴戊氧基)苯基]-10,15,20-三苯基卟啉(2b)的合成
取250mL已经干燥完全的三口烧瓶,加入0.096g(0.15mmol)单羟基卟啉,0.165g氢氧化钠,100mL无水THF,装好回流冷凝装置,氮气保护,在磁力搅拌下进行加热回流15min,溶液的颜色会变为绿色,用注射器加入1.3g(5.6mmol)1,5-二溴戍烷,加热回流至完全反应.将反应液倒入水中,用二氯甲烷进行三次萃取,然后将有机相合并,旋干得到粗产品.将此固体过硅胶柱分离,洗脱剂为二氯甲烷∶石油醚=1∶2,收集第一带旋干,得到紫色晶体0.061g,收率52%.
用类似方法合成2a和2c
化合物2a:紫色晶体0.073g,收率54%;1H NMR(400MHz,CDCl3)δ(ppm):8.91-8.80(m,8H),8.22-8.18(m,6H),8.12(d,J=8.5 Hz,2H),7.70-7.73(m,9H),7.28(d,J=8.8 Hz,2H),4.29(t,J=5.9Hz,2H),3.63(t,J=6.5Hz,2H),2.27-2.22(m,2H),2.17-2.04(m,2H),-2.78(s,2H).MS,m/z:764.22(M+).
化合物2b:紫色晶体0.061g,收率52%;1H NMR(400MHz,CDCl3)δ(ppm):8.92-8.77(m,8H),8.23-8.1(m,6H),8.11(d,J=8.5 Hz,2H),7.85-7.61(m,9H),7.27(d,J=8.8 Hz,2H),4.26(t,J=6.3Hz,2H),3.54(t,J=6.8Hz,2H),2.10-1.92(m,4H),1.83-1.78(m,2H),-2.78(s,2H).MS,m/z:778.23(M+).
化合物2c:紫色晶体0.083g,收率55%;1H NMR(400MHz,CDCl3)δ(ppm):8.89-8.84(m,8H),8.23-8.19(m,6H),8.10(d,J=8.4Hz,2H),7.79-8.73(m,9H),7.28(d,J=8.8Hz,2H),4.26(t,J=6.3Hz,2H),3.51(t,J=6.8Hz,2H),1.96-2.02(m,4H),1.69-1.63(m,4H),-2.78(s,2H).MS,m/z:792.25(M+).
1.3 4,4'-二羟基联苯尾饰卟啉(3)的合成
在两口烧瓶中加入0.024g(0.13mmol)联苯二酚,0.960g K2CO3,40mL无水DMF并在氮气保护下于90℃加热回流15min.用注射器注入0.103g(0.13mmol)用无水DMF溶解的化合物2b,100℃下加热回流2h.待反应液冷却后,倒入100mL水中,用乙酸乙酯萃取,并旋干萃取物.以石油醚∶二氯甲烷=3∶2为洗脱剂过硅胶柱收集第一带,旋干并称重.用类似方法合成3a和3c.
化合物3a:紫色晶体0.031g,收率44%;1H NMR(400MHz,CDCl3)δ(ppm):8.86-8.71(m,8H),8.16-8.14(m,6H),8.05(d,J=8.5 Hz,2H),7.73-7.62(m,9H),7.43(d,J=8.7 Hz,2H),7.37(d,J=8.6Hz,2H),7.22(d,J=8.6Hz,2H),6.96(d,J=8.7Hz,2H),6.81(d,J=8.6Hz,2H),4.28(t,J=5.2Hz,2H),4.14(t,J=5.4Hz,2H),2.16-2.08(m,4H),-2.85(s,2H).MS,m/z:908.31(M+).
化合物3b:紫色晶体0.054g,收率43%;1H NMR (400MHz,CDCl3)δ(ppm):8.92-8.82(m,8H),8.20-8.22(m,6H),8.12(d,J=8.5 Hz,2H),7.73-7.78(m,9H),7.48(d,J=8.7 Hz,2H),7.42(d,J=8.6Hz,2H),7.29(d,J=8.6Hz,2H),7.01(d,J=8.8Hz,2H),6.85(d,J=8.6Hz,2H),4.30(t,J=6.4Hz,2H),4.13(t,J=6.3Hz,2H),2.13-1.96(m,6H),-2.78(s,2H).MS,m/z:922.33(M+).
化合物3c:紫色晶体0.044g,收率47%;1H NMR(400MHz,CDCl3)δ(ppm):8.94-8.81(m,8H),8.25-8.19(m,6H),8.12(d,J=8.5 Hz,2H),7.78-7.73(m,9H),7.48(d,J=8.8 Hz,2H),7.46(d,J=8.8Hz,2H),7.28(d,J=8.6Hz,2H),7.00(d,J=8.8Hz,2H),6.91(d,J=8.8Hz,2H),4.28(t,J=6.4Hz,2H),4.18(t,J=6.9Hz,2H),4.09(t,J=6.4Hz,2H),3.96(t,J=6.4Hz,2H),2.06-2.02(m,2H),1.97-1.90(m,2H),-2.78(s,2H).MS,m/z:936.34(M+).
1.4 4,4'-二羟基联苯尾饰锌卟啉(4)的合成
取100mL单口瓶,加入0.030g(0.03mmol)化合物3b,0.7g醋酸锌,50mL二氯甲烷和3mL甲醇,安装好回流冷凝装置,磁力搅拌,加热回流至完全反应.将反应液倒入100mL水中,用100mL二氯甲烷进行三次萃取,将有机相合并,用无水硫酸镁进行干燥,旋干即可得到粗产品.将此固体产物过硅胶柱进行分离,用二氯甲烷∶石油醚=5∶1作为洗脱剂,收集第一带并旋干,得到了紫色晶体0.029 g,收率93%.
用类似方法合成4a和4c.
化合物4a:紫色晶体0.030g,收率94%;1H NMR(400MHz,CDCl3)δ(ppm):8.87-8.93(m,8H),8.20-8.10(m,6H),8.05(d,J=8.5 Hz,2H),7.75-7.60(m,9H),7.43(d,J=8.7 Hz,2H),7.37(d,J=8.6Hz,2H),7.21(d,J=8.7Hz,2H),6.96(d,J=8.7Hz,2H),6.81(d,J=8.6Hz,2H),4.29(t,J=5.4Hz,2H),4.14(t,J=5.3Hz,2H),2.17-2.09(m,4H).MS,m/z:970.23(M+).
化合物4b:紫色晶体0.029g,收率93%;1H NMR (400MHz,CDCl3)δ(ppm):8.87-8.93(m,8H),8.19-8.14(m,6H),8.05(d,J=8.5 Hz,2H),7.73-7.62(m,9H),7.41(d,J=8.7 Hz,3H),7.35(d,J=8.7Hz,2H),7.22(d,J=8.8Hz,2H),6.94(d,J=8.8Hz,2H),6.77(d,J=8.6Hz,2H),4.24(t,J=6.3Hz,2H),4.06(t,J=6.3Hz,2H),1.93-2.00(m,4H),1.81-1.77(m,2H).MS,m/z:984.24(M+).
化合物4c:紫色晶体0.045g,收率94%;1H NMR (400MHz,CDCl3)δ(ppm):8.93-8.87(m,8H),8.17-8.14(m,6H),8.04(d,J=8.5 Hz,2H),7.73-7.64(m,9H),7.39(d,J=8.7 Hz,2H),7.33(d,J=8.6Hz,2H),7.21(d,J=8.8Hz,2H),6.92(d,J=8.7Hz,2H),6.74(d,J=8.6Hz,2H),4.21(t,J=6.4Hz,2H),4.18-4.09(m,2H),4.01(t,J=6.4Hz,2H),2.02-1.90(m,4H),1.90-1.84(m,2H).MS,m/z:998.26(M+).
2 结果与讨论
2.1 化合物3和4的紫外-可见光谱研究
化合物3a~3c以及4a~4c的紫外-可见光谱如下图2所示.

图2 化合物3和4紫外-可见光谱图Fig.2 UV-vis of porphyrin 3 and 4 (in CH2Cl2)
化合物3a~3c以及4a~4c的紫外-可见光谱数据与四苯基卟啉(TPP)的紫外吸收光谱数据进行了比较与总结,如表1所示.

表1 化合物TPP及3a~4c紫外应谱数据Tab.1 UV-vis data of compounds TPP and 3a-4c(in CH2Cl2)
从表1可以看出,各个化合物均表现出了卟啉的特征吸收.与四苯基卟啉的紫外吸收光谱相比,化合物3和4的Soret带发生了1~2nm的红移,Q带发生了1~2nm的蓝移或者1~2nm红移.这说明联苯二酚基团的引入对卟啉化合物的吸收光谱产生了影响,但并不明显.根据Jean等[16]的理论可知:当卟啉分子的扭曲程度和分子共轭体系增大后,最低电子未占据轨道与最高电子占据轨道之间的能隙差减小,激发电子所需要的能量减小,电子在分子内很容易实现从基态到激发态的跃迁,从而导致卟啉的紫外吸收峰发生红移.因此,我们可以推测当把联苯二酚基团引入到卟啉分子后,整个卟啉的扭曲程度增大,并且改变了π电子的共轭程度,体系的HOMO与LUMO之间的能隙变窄,从而使得化合物的吸收峰发生红移.此外,金属卟啉的紫外吸收带也受中心金属离子的影响,锌离子的最外层电子结构是d10的满壳层状态,不含有未成对的电子结构,但在与卟啉环内的N原子发生配位后,使得N原子上本来不参与π-π*跃迁的孤对电子发生了变化,其中一个电子会直接参与键的形成,而另外的一个单电子则增加了共轭体系的π键的电荷密度,升高了成键轨道的能量,并且使能隙变窄,从而使得化合物的吸收光谱的位置发生了红移;同时S0→S1跃迁产生的Q带相应减少的原因是N原子与两个质子发生络合后,卟啉环内的4个N原子所处的化学环境变得趋于平均化,使得卟啉分子具有D4h的对称性,表现为QⅠ和QⅣ带的消失.锌离子与卟啉环配位后,锌离子突出于卟啉环平面的位置,从而影响了卟啉分子的平面性.锌离子与卟啉环内侧链基N原子间的相互弱作用,这些都是影响金属卟啉相对于未上金属的卟啉紫外吸收发生位移的重要因素.
2.2 化合物3和4的荧光光谱性质研究
化合物3a~3c以及4a~4c的荧光光谱如图3所示.

图3 化合物3和4荧光光谱图Fig.3 Fluorescence spectrum of porphyrin 3 and 4(in CH2Cl2)
化合物3a~3c以及4a~4c的荧光光谱数据与四苯基卟啉(TPP)的荧光光谱数据进行了比较与总结,如表2所示.
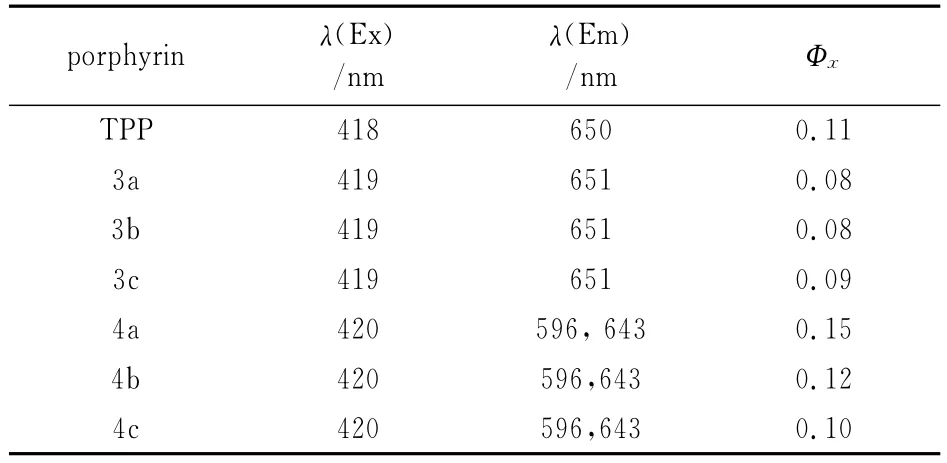
表2 化合物TPP与3a~4c荧光光谱数据Tab.2 Fluorencence spectral data of compounds TPP and 3a-4c(in CH2Cl2)
根据表2所列数据可知,联苯二酚修饰的锌卟啉的荧光量子产率要高于其卟啉化合物.原因是卟啉与锌离子配位后,增加了卟啉环间π-π作用,增大了卟啉环的刚性结构,减少了分子内能量的震动消耗,提高了能量转移效率.
本文合成的联苯二酚尾饰部分及其锌卟啉的最大荧光发射波长在650nm左右,位于可见的红光区,在可见光的红光部分有较强的荧光发射,这与理想的PDT药物应该具有的光学性质一致,有望成为一类优良的光敏剂.
[1]LI G T,BHOSALE S V,WANG T,et al.Nanowells on silica particles in water containing long-distance porphyrin heterodimers[J].J Am Chem Soc,2003,125(35):10693-10702.
[2]YOU C C,WURTHNER F.Porphyrin-perylene bisimide dyads and triads:synthesis and optical and coordination properties[J].Organic Letters,2004,6(14):2401-2404.
[3]SPRINGER J,KODIS G,GARZA L,et al.Stepwise sequential and parallel photoinduced charge separation in a porphyrintriquinone tetrad[J].J Phys Chem A,2003,107(18):3567-3575.
[4]LAU R L C,JIANG J Z,DENNIS K P,et al.Fourier transform ion cyclotron resonance studies of lanthanide(III)porphyrin-phthalocyanine heteroleptic sandwich complexes by using electrospray ionization[J].Journal of American Society for Mass Spectrom,1997,8(2):161-169.
[5]POLICARD A,LEULLER A.Etudesur les aspects offers par des tumeurs experimentales examineles a la luminere de woods[J].Compte Rendus Soc biol,1924,91(1):1423-1424.
[6]ADLER A D,LONGO F R,SHERGALIS W.Mechanistic investigations of porphyrin syntheses(I)preliminary studies on ms-tetraphenylporphin[J].Journal of the American Chemical Society,1964,86(15):3145-3149.
[7]ERIKSSON E S E,ERIKSSON L A P,NAKAGAWA K,et al.An optochemical HCl gas sensor using 5,10,15,20-tetrakis(3′,5′-di-tert-butyl,4′-hydroxyphenyl)porphin-ethylcellulose composite films[J].Sensors and Actuators B:Chemical,1998,52(1/2):10-14.
[8]SESSLER J L,WEGHORN S J.Expanded,contracted &isomeric porphyrins[M].New York:Elsevier Scienced Ltd,1997:392-394.
[9]BRUNNER H,GRUBER N.Carboplatin-containing porphyrinplatinum complexes as cytotoxic and phototoxic antitumor agents[J].Inorganica Chimica Acta,2004,357(15):4423-4451.
[10]LI Dong-hong,LIU Jian-cang,YU Ke-gui,et al.Study on the antitumor activity of anticancer drugs porphyrin modified[C]//The Fifth National Conference on Chemical Biology.2007:121.
[11]MAGDA D J,WEI W H,WANG Z,et al.Synthesis of texaphyrin conjugates[J].Pure and Applied Chemistry,2004,76(2):365-374.
[12]贾志云,邓侯富.血卟啉类化合物在肿瘤诊疗应用的研究进展[J].中国医药工业杂志,2006,37(6):426-430.JIA Zhi-yun,DENG Hou-fu.Progress of applications of hematoporphyrin derivatives on diagnosis and treatment of tumor[J].Chinese Journal of Pharmaceuticals,2006,37(6):426-430.(In Chinese)
[13]VENTURINI M.Rational development of capecitabine[J].European Journal of Cancer,2002,38(2):3-9.
[14]PAZDUR R,HOFF P M,MEDGYESY D,et al.The oral fluorouracil prodrugs[J].Oncology (Huntingt),1998,10(7):48-51.
[15]MARCUCCIO S M,ELMES B C,HOLAN G,et al.Modified nucleosides.II.1economical synthesis of 2′,3′-dideoxycytidine[J].Nucleosides and Nucleotides,1992,11(10):1695-1701.
[16]JEAN B K,JOHN J L,FREDERICK R L.A mechanistic study of the synthesis and spectral properties of meso-tetraaryl-porphyrins[J].Journal of the American Chemical Society,1972,94(11):3986-3992.
猜你喜欢
化工学报(2021年1期)2021-01-30
农药科学与管理(2019年8期)2019-11-23
山东化工(2019年2期)2019-02-16
福建农林大学学报(自然科学版)(2018年5期)2018-10-11
科技知识动漫(2017年7期)2017-08-09
化学工业与工程(2015年1期)2015-02-10
中国药理学通报(2014年2期)2014-05-09
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年7期)2014-02-28
cookie world(2010年2期)2010-03-04
