
EphB4基因重组慢病毒载体的构建及在结肠癌细胞中的表达
2014-09-28 06:29:14夏秋媛王建东周晓军
医学研究生学报 2014年9期
张 锦,夏秋媛,王建东,周晓军
0 引 言
EphB4是受体酪氨酸激酶家族(receptor tyrosine kinases)中最大的亚群——促红细胞生成素产生肝细胞受体中的一员。与其他Eph分子相比,EphB4具有重要而独特的生物学功能。研究表明,EphB4与其配体EphrinB2相结合在血管及淋巴管发育、重塑等过程中发挥重要的调控作用[1-3]。同时,EphB4也可通过调控细胞增殖、分化、凋亡等过程参与众多肿瘤的发生发展[4-5]。但 EphB4在不同类型肿瘤或同种肿瘤的不同阶段发挥的作用并不相同,有时甚至完全相反[6]。为了更好地了解EphB4基因在结肠癌发生发展中的作用及其相关机制,为结肠癌的基因治疗提供有益的实验依据,本研究构建了含有EphB4基因全长片段的慢病毒载体,并进一步转染人结肠癌细胞株,检测转染前后细胞株内EphB4的表达变化。
1 材料与方法
1.1 材料 SW480、Caco-2购自中科院上海细胞库;pMD18-T vector购自Takara公司;感受态细胞DH5α、HEK293细胞、293T 细胞、platinum Pfx DNA Polymerase、platinum Taq DNA Polymerase High Fidelity、pcDNA3.1(+)、慢病毒载体 pLenti6.3/V5-DEST、pLenti6.3-MCS-IRES2-EGFP、包装质粒 pLP1、pLP2 及 pLP/VSVG、Packaging Mix、lipofectamine 2000、BP clonaseⅡ、LR clonaseⅡ、蛋白酶 K、pDONR221、Opti-MEM 培养液、Polybrene、DMEM 培养基、胎牛血清、Blasticidin、0.25%Trypsin、Trizol、SuperScripⅢ RT(200 U/μL)、RNase H等均购自Invitrogen公司;BamHⅠ、NotⅠ、AscⅠ购自 MBI公司;T4 DNA Ligase购自NEB公司。
1.2 方法
1.2.1 EphB4基因序列的合成 对所需合成的EphB4基因序列进行分析,检查基因内部有无复杂的二级结构和重复序列连接。根据基因序列分析的结果,分别设计合成一系列的单链oligo。利用PCR将合成的oligo拼接成完整的基因序列。将合成好的序列装入pMD18-T载体并转化至感受态细胞DH5α。测序验证重组克隆中插入序列是否与EphB4基因序列相一致。通过重叠PCR将基因序列中突变点修复,命名为pMD18-EphB4。
1.2.2 含有EphB4基因序列的质粒载体的构建 应用限制性内切酶 NotⅠ、BamHⅠ分别对 pMD18-EphB4和载体pcDNA3.1(+)进行双酶切,回收的基因片段和载体片段通过T4 DNA Ligase 16℃连接2h后转化至感受态细胞DH5α。测序验证重组克隆插入片段的序列信息。质粒命名为pcDNA3.1-EphB4。
1.2.3 慢病毒表达载体的构建 应用AscⅠ、BamHⅠ双酶切pcDNA3.1-EphB4质粒及慢病毒载体pLenti6.3-MCS-IRES2-EGFP,通过 T4 DNA Ligase将两者酶切后电泳回收的产物连接,并转化DH5α。测序验证重组克隆中插入片段的序列信息,并将其命名为 pLenti6.3-EphB4-IRES2-EGFP。
1.2.4 慢病毒的包装 取9 μg Packaging Mix和3 μg慢病毒表达质粒 pLenti6.3-EphB4-IRES2-EGFP加入 1.5 mL Opti-MEM,轻轻混匀后加入 lipofectamine 2000稀释液,室温孵育20 min制备质粒脂质体复合物。将复合物缓慢加入293T细胞,置于37℃、5%CO2的培养箱中培养6 h后,更换为完全培养液,继续培养48 h后收集细胞培养液上清,离心并过滤。将所获含有EphB4基因序列的病毒液命名为pLenti6.3-EphB4并进行滴度测定。
1.2.5 病毒滴度的测定 HEK293细胞经胰酶消化后轻轻吹打成单细胞悬液,细胞计数后按8000个细胞每孔的密度铺于96孔板。按文献[7]将病毒液用慢病毒稀释用培养基(DMEM+2%FBS+8 μg/mL Polybrene)进行1∶10倍比稀释后,各取100 μL感染96孔板中的HEK293细胞。37℃、5%CO2培养箱中培养24 h,更换成完全培养基继续培养96 h,在荧光显微镜下观察各孔中荧光细胞数量。每个稀释度2个重复。
病毒滴度=各孔中表达荧光的细胞数量平均值/每孔中含有的慢病毒液体积
1.2.6 慢病毒侵染靶细胞构建稳转株 靶细胞经胰酶消化、细胞计数后按照1.5×105个/孔的密度接种于6孔板中培养24 h,换成不含血清的培养基,分别加入感染复数为20的慢病毒Lenti6.3-EphB4和Lenti6.3-EGFP(阴性对照)稀释液继续培养。48 h后于荧光显微镜下观察绿色荧光蛋白(green fluorescene protein,GFP)的表达情况,并加入一定浓度的Blasticidin进行筛选,72 h后观察细胞情况,后续每4~72小时更换1次含有抗生素Blasticidin的完全培养基。待空白孔细胞完全死亡后,继续培养并适时冻存,同时收集部分细胞抽提RNA检测EphB4 mRNA表达。设定空白的靶细胞作为对照。
1.2.7 qPCR检测目的基因的表达情况 Trizol法提取各组细胞样品中总RNA并逆转录为cDNA。通过qPCR法检测细胞样品中目的基因EphB4、EphrinB2和内参基因GAPDH的表达量,实验重复3次。根据qPCR反应曲线得到各样品目的基因和内参基因的域值循环数(threshold cycle number,Ct值),采用ΔΔCt的方法对基因表达进行相对定量。以转染阴性对照载体的样品作为对照样品,比较各瞬时转染干扰载体组样品中目的基因的表达量,并计算干扰效果。计算公式如下:
ΔΔCt=(待测样品目的基因的Ct平均值-待测样品内参基因的Ct平均值)-(对照样品目的基因的Ct平均值-对照样本内参基因的Ct平均值)
基因的表达量 F=2-ΔΔCt
目的基因的干扰效率=1-2-ΔΔCt
目的基因EphB4、EphrinB2及内参基因GAPDH的引物序列如下:EphB4-F:5'-CGCACCTACGAAGTGTGTGA-3',EphB4-R:5'-GTCCGCATCGCTCTCATAGTA-3';GAPDH-F:5'-GAAGGTCGGAGTCAACGGATT-3',GAPDH-R:5'-CGCTCCTGGAAGATGGTGAT-3'。PCR 反 应条件为95℃ 2min;95℃ 10s,60℃ 30s,70℃ 45s,40 个循环;95℃ 30s。每个样本重复检测3次。
2 结 果
2.1 EphB4基因序列的合成及鉴定 人工化学合成寡核苷酸片段,通过片段连接技术将系列小片段连接成EphB4基因,装入pcDNA3.1(+)载体进行扩增后,双向测序结果显示合成的EphB4序列与Gene Bank中公布的序列一致,表明成功获得了2964 bp的EphB4基因。
2.2 慢病毒载体pLenti6.3-EphB4-IRES2-EGFP的成功构建 通过酶切及连接反应将质粒pcDNA3.1-EphB4中扩增的目的基因EphB4片段克隆装入慢病毒表达载体pLenti6.3-MCS-IRES2-EGFP,构建含有EphB4基因序列的慢病毒表达载体。经过双向测序,证实重组克隆中插入片段的序列信息与EphB4基因序列一致,慢病毒表达载体构建成功。
2.3 含有EphB4基因序列的慢病毒的获得及滴度测定 慢病毒表达载体 pLenti6.3-EphB4-IRES2-EGFP与包装质粒pLP1、pLP2及pLP/VSVG通过脂质体介导获得了含有EphB4基因序列的慢病毒Lenti6.3-EphB4。通过稀释计数法测得病毒滴度为1×108TU/mL。
2.4 稳转细胞株的获得及评价 病毒液Lenti6.3-EphB4和Lenti6.3-EGFP侵染靶细胞SW480及Coca-2后,根据本实验所用的慢病毒载体中所含的抗性标志选用药物Blasticidin对靶细胞进行筛选及维持,最终获得了稳定的细胞系。荧光显微镜下观察病毒转染后72 h的各组细胞均可见GFP表达。见图1。qPCR检测证实,与原始SW480或Caco-2细胞相比,Lenti6.3-EphB4侵染的SW480和Caco-2中EphB4表达显著增加,而Lenti6.3-EGFP病毒转染的SW480和Caco-2细胞中EphB4表达未见明显变化。见图2。
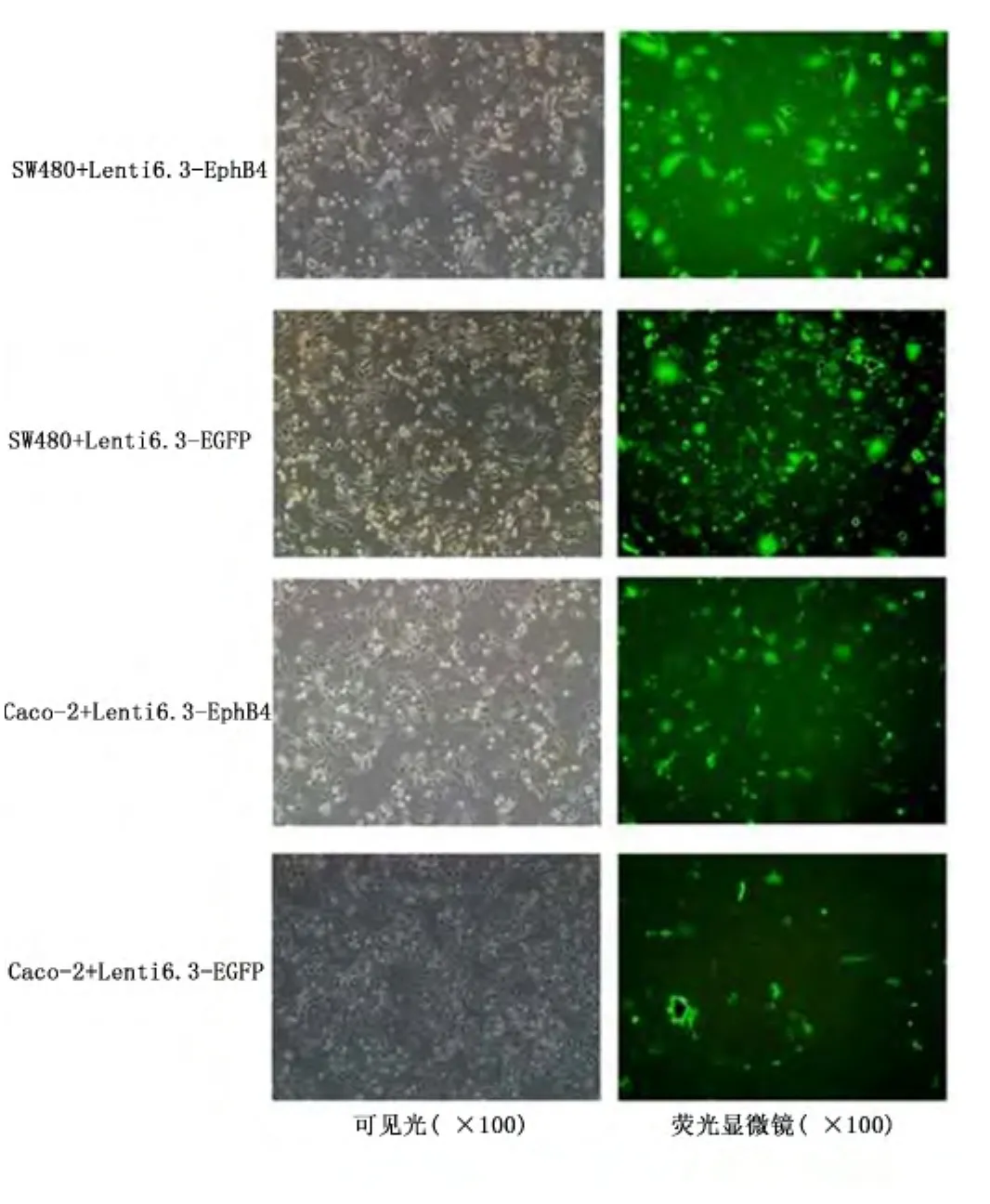
图1 稳定过表达EphB4的结肠癌细胞株Figure 1 Colon cancer cell lines stably overexpressing EphB4
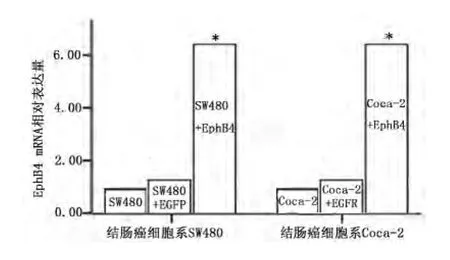
图2 各组细胞株EphB4 mRNA的相对含量Figure 2 Relative expression of EphB4 mRNA in different colon cancer cell lines
3 讨 论
近年来对恶性肿瘤发生发展分子病理机制的深入研究为正在兴起的基因治疗提供了理论依据,促进了恶性肿瘤分子靶向治疗的探索性研究。作为一种可能的分子治疗新靶点,明确EphB4的具体功能及作用机制尤为重要。而新近分子生物学技术的发展为实现上述研究目的提供了可能[8]。本研究证实了经外源性手段(如慢病毒载体介导)将人工合成的基因有效地整合到宿主染色体上,从而达到持久性表达的可能性,为靶向EphB4基因治疗提供了实验依据。
实验中选用的慢病毒载体包含了包装、转染、稳定整合所需要的遗传信息[9]。慢病毒包装质粒提供所有的转录及包装所需要的所有辅助蛋白。利用表达载体和包装质粒同时共转染细胞,在细胞中进行病毒的包装,包装好的假病毒颗粒分泌到细胞外的培养基中,产生高滴度的病毒颗粒,可以直接用于宿主细胞的感染。目的基因进入到宿主细胞之后,经过反转录,整合到基因组,从而高水平的表达效应分子。为了获得稳定过表达目的基因的细胞系,本研究进一步运用基因转染方式把抗药性基因引入细胞,并确定抗药性基因跟需要筛选的目标基因受同样的启动子调控,然后将相应的药物加入到培养基,不表达目标基因和抗药性基因的细胞将全部被药物杀死,从而达到筛选的目的,获得高度纯化的阳性转染细胞[10-11]。与通过流式细胞仪分选携带GFP阳性细胞的方法相比,此方法操作起来更为简便易行,因此也是目前筛选稳转细胞株最常用的方法。
区别于一般的逆转录病毒载体或其他非病毒载体,慢病毒载体对分裂细胞和非分裂细胞均具有感染能力。研究发现,慢病毒载体转移基因片段容量大,不易诱发宿主免疫反应,能稳定整合于靶细胞的基因组;同时慢病毒在整合基因时,并不倾向于转录的起始位点,而是在基因的整个转录区都可以整合,整合的慢病毒对启动子的激活概率更低。与其他致癌反转录病毒载体相比,慢病毒载体不易引起插入突变[12-14]。因此本研究选用了慢病毒作为载体将外源性基因EphB4转导入靶细胞。转染24h及72h后,荧光显微镜下转染含有随机序列及转染EphB4基因的慢病毒载体的各组细胞中均见到GFP表达,且与原始细胞相比,转染慢病毒载体的各组细胞细胞形态未见明显变化,qPCR实验证实相对于原始细胞,含有EphB4基因序列的慢病毒转染的靶细胞中EphB4基因的表达量明显升高。
综上所述,本研究成功运用慢病毒为载体,将人工合成的EphB4基因序列转导入靶细胞,并通过靶细胞抗生素筛选实验,最终获得了稳定过表达EphB4基因的结肠癌细胞株,对于研究结肠癌的基因治疗提供了有益的实验依据。
[1]Klein R.Eph/ephrin signalling during development[J].Development,2012,139(22):4105-4109.
[2]Nakamoto M,Bergemann AD.Diverse roles for the Eph family of receptor tyrosine kinases in carcinogenesis[J].Microsc Res Tech,2002,59(1):58-67.
[3]戴颖青,张 秦,刘 琦.Eph受体在卵巢癌中的研究进展[J].医学研究生学报,2012,25(10):1112-1115.
[4]郭 庆,王建东,石群立.受体酪氨酸激酶EphA2在肾细胞癌发生及发展中的作用[J].医学研究生学报,2012,25(12):1333-1336.
[5]Salvucci O,Tosato G.Essential roles of EphB receptors and EphrinB ligands in endothelial cell function and angiogenesis[J].Adv Cancer Res,2012,114:21-57.
[6]Noren NK,Pasquale EB.Paradoxes of the EphB4 receptor in cancer[J].Cancer Res,2007,67(9):3994-3997.
[7]Boden D,Pusch O,Silbermann R,et al.Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins[J].Nucleic Acids Res,2004,32(3):1154-1158.
[8]Attwood BK,Patel S,Pawlak R.Ephs and ephrins:emerging therapeutic targets in neuropathology[J].Int J Biochem Cell Biol,2012,44(4):578-581.
[9]D'Costa J,Mansfield SG,Humeau LM.Lentiviral vectors in clinical trials:Current status[J].Curr Opin Mol Ther,2009,11(5):554-564.
[10]Sakuma T,Barry MA,Ikeda Y.Lentiviral vectors:basic to translational[J].Biochem J,2012,443(3):603-618.
[11]Stewart HJ,Fong-Wong L,Strickland I,et al.A stable producer cell line for the manufacture of a lentiviral vector for gene therapy of Parkinson's disease[J].Hum Gene Ther,2011,22(3):357-369.
[12]陈 伟,曹 罡,董 震,等.人Notch4基因RNAi慢病毒载体的构建及鉴定[J].医学研究生学报,2013,26(2):116-121.
[13]Dissen GA,Lomniczi A,Neff TL,et al.In vivo manipulation of gene expression in non-human primates using lentiviral vectors as delivery vehicles[J].Methods,2009,49(1):70-77.
[14]Guy HM,McCloskey L,Lye GJ,et al.Characterization of lentiviral vector production using microwell suspension cultures of HEK293T-derived producer cells[J].Hum Gene Ther Methods,2013,24(2):125-139.
猜你喜欢
新传奇(2022年51期)2023-01-04 21:51:14
分子诊断与治疗杂志(2022年11期)2022-12-23 13:13:18
中国免疫学杂志(2019年3期)2019-03-13 02:11:14
神州·中旬刊(2019年1期)2019-02-12 08:47:44
西南国防医药(2016年7期)2016-12-01 06:01:15
中国卫生标准管理(2015年1期)2016-01-14 03:41:26
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国当代医药(2015年16期)2015-03-01 02:03:11
中国医药导报(2015年27期)2015-02-28 22:08:02
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
