
苯并呋喃与苯并二氧六环类新木脂素及其衍生物的合成与生物活性研究*
2014-09-18 02:52汪秋安余玲敏刘双艳
湖南大学学报(自然科学版) 2014年7期
汪秋安,徐 雨,余玲敏,刘双艳
(湖南大学 化学化工学院,湖南 长沙 410082)
苯并呋喃和苯并二氢呋喃新木脂素类是存在于丹参、百部、龙血巴豆、西洋参、野花椒、水飞蓟、牛蒡子等药用植物中的天然有机化合物,它们具有良好的生物活性,如抗病毒、抗肿瘤、抗菌、抗氧化、免疫抑制剂、抗血小板聚集活性和神经营养作用等[1-5].例如:从南美洲大戟科龙血巴豆树茎中分离出来的苯并呋喃新木脂素3′,4-di-O-methylcedrusin具有良好的抗肿瘤活性[6].从羊角草中分离得到的苯并二氧六环新木脂素cleomiscosinde A也具有显著的抗肿瘤活性[7].从美洲商陆Phytolacca americana L种子中分离得到的苯并二氧六环新木脂素isoamericanol A,具有营养神经的活性,可提高胎鼠大脑半球胆碱乙酰转移酶的活性,改善神经条的形态[8].
为了研究这类化合物的生理活性和构效关系,以及新药开发的需要,我们探索了简便高效的合成苯并呋喃类和苯并二氧六环类新木脂素的方法,并进一步研究这些化合物的生理活性.以3,4-二羟基苯丙烯酸(咖啡酸)为原料,以仿生氧化偶联和DDQ脱氢反应为关键步骤,合成了一系列苯并呋喃新木脂素类化合物1,3~7和苯并二氧六环新木脂素类化合物2,8~10.其中5~7,9和10是未见文献报道的新化合物,8为天然产物美洲商陆醇A合成路线如图1所示.
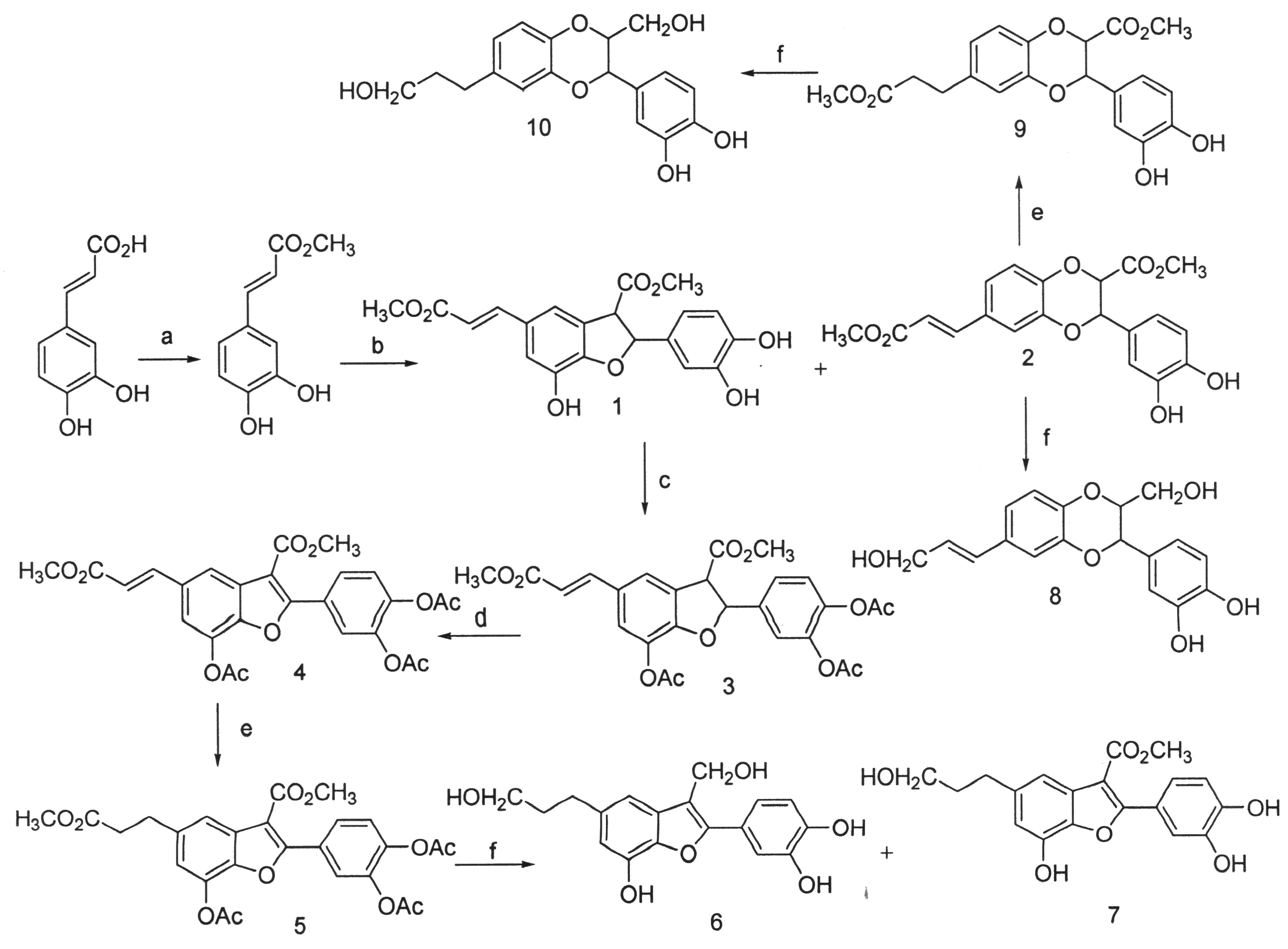
Reagent and conditions:(a) MeOH,concentrated sulfuric acid,reflux; (b) Ag2O,anhydrous toluene,anhydrous acetone,r.t,dark; (c) Ac2O,pyridine,r.t; (d) DDQ,1,4-dioxane,reflux,48h; (e) 10% Pd-C,H2,THF,r.t;(f) LiAlH4,anhydrous THF,-20 oC→r.t.
1 实验部分
1.1 仪器与试剂
核磁共振仪:Bruker-AV400,400 MHz(各种氘代溶剂,TMS为内标);质谱(ESI)用VG Autospec-3000,SHIMADZ qp-500;红外光谱用Bruker Tensor-27(KBr压片法);熔点用XRC-1型显微熔点仪测定(温度未校正).所用试剂如无特殊说明均为市售化学纯或者分析纯;柱层析用硅胶300~400目(青岛海洋化工厂产品).3,4-二羟基苯基丙烯酸甲酯按文献[9]合成.
1.2 2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-羟基-2,3-二氢苯并呋喃(1)和2-(3',4′-二羟基苯基)-3-甲氧羰基-7-甲氧羰基乙烯基-2,3-二氢-1,4-苯并二氧六环(2)的合成
在100 mL的三颈圆底烧瓶中加入化合物32.5 g(12.88 mmol)和新制氧化银粉末1.99 g(8.59 mmol),再加入无水丙酮20 mL,无水甲苯40 mL.在N2保护下室温避光搅拌,TLC监测反应终点.约48 h后停止反应,过滤,用丙酮洗涤,减压旋除溶剂,得红褐色黏稠物.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)= 4∶1~3∶1],分别得20.9 g 和10.96 g,产率分别为36%和39%.
化合物1:黄色黏稠物.1H NMR (400 MHz,CDCl3)δ(ppm):7.56 (1H,d,J=16.0 Hz,8-H),7.04 (1H,s,4-H),6.99 (1H,s,6-H),6.84 (1H,d,J=2.0 Hz,2′-H),6.79 (1H,d,J=8.0 Hz,5′-H),6.76 (1H,dd,J=8.0,2.0 Hz 6′-H),6.26 (1H,d,J=16.0 Hz,9-H),6.01 (1H,d,J=7.6 Hz,2-H),4.26 (1H,d,J=7.6 Hz,3-H),3.80 (3H,s,10-OCH3),3.78 (3H,s,11-OCH3); MS (ESI)m/z:387 [M+H]+.
化合物2:黄色油状物.1H NMR (400 MHz,CDCl3)δ(ppm):7.58 (1H,d,J=16.0 Hz,8-H),7.14 (1H,s,5-H),7.09 (1H,s,6-H),6.98 (1H,s,7-H),6.96 (1H,s,2′-H),6.85 (1H,s,5′-H),6.77 (1H,s,6′-H),6.59 (1H,s,3′-OH),6.46 (1H,s,4′-OH),6.29 (1H,d,J=16.0 Hz,9-H),5.08 (1H,d,J=6.4 Hz,2-H),4.67 (1H,d,J=6.4 Hz,3-H),3.79 (3H,s,10-OCH3),3.64 (3H,s,11-OCH3); MS(ESI)m/z:387 [M+H]+.
1.3 2-(3′,4′-乙酰氧基苯基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-乙酰氧基-2,3-二氢苯并呋喃(3)的合成
在100 mL的单口烧瓶中加入化合物1 238 mg(0.617 mmol),加入无水吡啶10 mL将其溶解.室温搅拌10 min后,加入乙酸酐5 mL,TLC监测反应,直至原料点消失.约3 h后停止反应,将反应液倾入装有30 mL冰水的烧杯中搅拌20 min,出现白色浑浊,用乙酸乙酯(3×20 mL)萃取,合并有机相依次用5%稀盐酸、饱和食盐水洗涤,最后用无水硫酸镁粉末干燥.蒸除溶剂得黄色固状物,用甲醇重结晶后得白色膨松固体285 mg,产率90%.m.p.127-128oC;1H NMR (400 MHz,CDCl3)δ(ppm):7.63 (1H,d,J=15.6 Hz ,8-H),7.44 (1H,s,4-H),7.30 (1H,dd,J=8.4,2.0 Hz 6′-H),7.24 (1H,d,J=2.0 Hz,2′-H),7.21 (1H,d,J=8.4 Hz,5'-H),7.19 (1H,s,6-H),6.33 (1H,d,J=16.0 Hz,9-H),6.22 (1H,d,J=7.6 Hz,2-H),4.32 (1H,d,J=7.6 Hz,3-H),3.85 (3H,s,10-OCH3),3.79 (3H,s,11-OCH3),2.32 (3H,s,7-OCH3),2.28 (6H,s,3′-COCH3,4'-COCH3); MS (ESI)m/z:513 [M+H]+.
1.4 2-(3',4'-乙 酰 氧 基 苯 基)-3-甲氧羰基-5-甲氧羰基乙烯基-7-乙 酰氧基苯并呋喃(4)的合成
在100 mL三颈烧瓶内加入化合物3 500 mg(0.976 mmol),DDQ(2,3-二氯-5,6-二氰基-1,4-苯醌 )2 g(9.76 mmol),在N2氛围下加入无水1,4-二氧六环30 mL,继续通入N25 min,换无水氯化钙干燥管.加热回流,TLC监测反应,直至原料点基本消失.48 h后停止反应,将反应液冷却,过滤,滤液中倒入溶有NaHSO33.05 g(29.33 mmol)的水溶液100 mL,CH2Cl2200 mL,充分搅拌后分液.水层用二氯甲烷萃取(3×20 mL),有机相合并用饱和食盐水洗涤,无水Na2SO4干燥.硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=3∶1],得白色固体315 mg,产率63%.m.p.157-158oC;1H NMR (400 MHz,DMSO-d6)δ(ppm):8.20(1H,s,4-H),7.93 (1H,s,6-H),7.91 (1H,dd,J=2.4,8.0 Hz,6'-H),7.82 (1H,d,J=8.0 Hz,5′-H),7.81 (1H,s,2′-H),7.50 (1H,d,J=16.4 Hz,8-H),6.74 (1H,d,J=16.4 Hz,9-H ),3.91 (3H,s,10-OCH3),3.75 (3H,s,11-OCH3),2.43 (3H,s,7-COCH3),2.33 (6H,s,3′-COCH3,4′-COCH3).MS (ESI)m/z:511 [M+H]+.
1.5 2-(3′,4′-乙酰氧基苯基)-3-甲氧羰基-5-甲氧羰基乙基-7-乙酰氧基苯并呋喃(5)的合成
在100 mL的单口圆底烧瓶内加入化合物4100 mg(0.196 mmol),5% Pd-C 40 mg,加入无水THF 15 mL.氢气氛围下室温磁力搅拌12 h,TLC监测反应至原料点很浅,过滤,减压旋干溶剂.湿法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=3∶1],得白色固体92 mg,产率92%.m.p.152-153oC;1H NMR (400 MHz,CDCl3)δ(ppm):7.82 (1H,s,4-H),7.68 (1H,s,6-H),7.23 (1H,dd,J=2.4,8.0 Hz,6′-H),7.19 (1H,s,2′-H),6.91 (1H,s,5′-H),3.86 (3H,s,10-OCH3),3.61 (3H,s,11-OCH3),3.00 (2H,t,J=8.0 Hz,8-CH2-),2.62 (2H,t,J=8.4 Hz,9-CH2-),2.33 (3H,s,7-COCH3),2.25 (6H,s,3′-COCH3,4′-COCH3).MS(ESI)m/z:513 [M+H]+.
1.6 2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃(6)和2- (3',4'-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃(7)的合成
称取四氢铝锂47.5 mg(1.25 mmol)于100 mL单口瓶中,加入10 mL无水乙醚.于-20oC,N2氛围下将溶有化合物580 mg(0.156 mmol)的12 mL无水THF缓慢滴加至其中.在-20oC下磁力搅拌反应,反应2 h后,改室温反应,TLC监测直至原料点消失.缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×12 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)/V(甲醇)=15∶15∶1],得无色油状物6 32 mg和无色油状物7 25 mg,经表征,无色油状物6为2-(3′,4′-二羟基苯基)-3-羟基甲基-5-羟基甲基乙基-7-羟基苯并呋喃,产率57%.1H NMR(400 MHz,DMSO-d6)δ(ppm):9.07 (1H,s,7-OH),8.96 (1H,s,3′-OH),8.89 (1H,s,4′-OH),6.73 (1H,s,2′-H),6.67 (1H,d,J=8.0 Hz,5′-H),6.61 (1H,dd,J=2.0,8.0 Hz ,6′-H),6.52 (1H,s,6-H),6.47 (1H,s,4-H),5.00 (1H,s,10-OH),4.45 (1H,s,11-OH),3.30 (2H,t,J=8.0 Hz,10-CH2-),3.16 (2H,s,11-CH2-),2.44 (2H,t,J=8.0 Hz,8-CH2-),1.62-1.66 (2H,m,9-CH2-); MS(ESI) m/z:331 [M+H]+.
无色油状物7为2-(3′,4′-二羟基苯基)-3-甲氧羰基-5-羟基甲基乙基-7-羟基苯并呋喃,产率49%.1H NMR(400 MHz,DMSO-d6)δ(ppm):10.14 (1H,s,7-OH),9.62 (1H,s,3′-OH),9.33 (1H,s,4′-OH),7.44 (1H,s,2′-H),7.36 (1H,dd,J=2.0,8.0 Hz ,6′-H),7.17 (1H,s,6-H),6.87 (1H,d,J=8.4 Hz,5′-H),6.64 (1H,s,4-H), 4.50 (1H,s,10-OH),3.85 (3H,s,11-OCH3),3.43 (2H,t,J=7.6 Hz,10-CH2-),2.62 (2H,t,J=8.0 Hz,8-CH2-),1.70-1.74 (2H,m,9-CH2-); MS(ESI)m/z:359 [M+H]+.
1.7 2-(3′,4′-二羟基苯基)-3-羟基甲基-7-羟基甲基乙烯基-2,3-二氢-1,4-苯并二氧六环(8)的合成
在50 mL的单口圆底瓶中加入四氢铝锂12 mg(0.31 mmol),无水四氢呋喃10 mL 于-20oC,N2氛围下将溶有化合物240 mg(0.11 mmol)的10 mL无水四氢呋喃缓慢滴加至其中.在-20oC下磁力搅拌反应1 h,撤出低温,室温过夜反应,TLC监测至原料点消失,反应液呈灰绿色.缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×15 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.过滤旋干溶剂,干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=1∶1],得白色固体21 mg,产率59%.m.p.147-149oC [文献值[10]:147-150 ℃];1H NMR (400 MHz,DMSO-d6)δ(ppm):8.90 (3H,s,3′-OH,4′-OH,11-OH),7.73 (1H,d,J=15.8 Hz,8-H),6.92 (2H,s,5-H,6-H),6.71 (2H,s,5′-H,6′-H),6.68 (1H,s,2′-H),6.61 (1H,s,7-H),6.47 (1H,d,J=16.0 Hz,9-H),5.41 (1H,d,J=5.6 Hz,2-H),5.04 (1H,s,10-OH),4.77-4.79 (1H,m,3-H),4.08 (2H,d,J=4.4 Hz,11-CH2-),4.02 (2H,d,J=6.4 Hz,10-CH2-).MS(ESI)m/z:331 [M+H]+.
1.8 2-(3′,4′-二羟基苯基)-3-甲氧羰基-7-甲氧羰基乙基-2,3-二氢-1,4-苯并二氧六环(9)的合成
在100 mL的圆底烧瓶中加入化合物2 530 mg(1.37 mmol),5% Pd/C 200 mg,然后加入无水THF 20 mL.氢气氛围下室温磁力搅拌12 h,TLC监测反应至原料点消失,过滤,减压蒸除溶剂.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=4∶1],得无色油状物489 mg,产率92%.1H NMR (400 MHz,CDCl3)δ(ppm):6.89 (1H,s,5-H),6.85 (1H,s,6-H),6.81 (1H,s,7-H),6.75 (1H,s,2′-H),6.70 (1H,s,3′-H),6.68 (1H,s,4′-H),5.10-5.80 (2H,s,3′-OH,4′-OH),5.00 (1H,d,J=6.8 Hz,2-H),4.58 (1H,d,J=6.0 Hz,3-H),3.66 (3H,s,10-OCH3),3.61 (3H,s,11-OCH3),2.84 (2H,t,J=7.6 Hz,8-CH2-),2.59 (2H,t,J=8.0 Hz,9-CH2-).MS(ESI) m/z:389 [M+H]+.
1.9 2-(3′,4′-二羟基苯基)-3-羟基甲基-7-羟基丙基-2,3-二氢-1,4-苯并二氧六环(10)的合成
在100 mL的单口瓶中加入四氢铝锂147 mg(3.86 mmol),加入无水乙醚10 mL于-20oC,N2氛围下将溶有化合物9 300 mg(0.77 mmol)的12 mL无水THF缓慢滴加至其中.在-20oC下磁力搅拌反应,反应4 h后,TLC监测原料点消失.将反应撤至室温,缓慢滴加稀盐酸淬灭反应,用乙酸乙酯萃取(3×15 mL),合并有机相用饱和食盐水洗涤,无水Na2SO4干燥.干法上样,硅胶柱层析分离[洗脱剂:V(石油醚)/V(乙酸乙酯)=4∶1],得无色油状物132 mg,产率51%.1H NMR(400 MHz,DMSO-d6)δ(ppm):9.05 (1H,s,3′-OH),9.01 (1H,s,4′-OH),6.80 (1H,s,2′-H),6.78 (1H,d,7-H),6.73 (1H,s,5-H),6.70 (1H,d,6-H),6.67 (1H,d,5′-H),6.65 (1H,d,6′-H),4.90 (1H,s,11-OH),4.79 (1H,d,J=7.6 Hz,2-H),4.34 (1H,s,10-OH),3.96 (1H,d,J=7.2 Hz,3-H),3.51 (2H,d,J=13.6 Hz,11-CH2-),3.37-3.40 (2H,m,10-CH2-),2.34 (2H,t,J=8.0 Hz,8-CH2-),1.62-1.69 (2H,m,9-CH2-); MS(ESI)m/z:333 [M+H]+.
1.10 生物活性测试
实验方法:1)接种细胞:用含10%胎牛血清的培养液(DMEM或者RMPI1640)配成单个细胞悬液,以每孔5 000~10 000个细胞接种到96孔板,每孔体积100 μL,贴壁细胞需提前12 h接种培养.
2)加入待测化合物溶液(固定浓度40 μM初筛,于该浓度对肿瘤细胞生长抑制在50%附近的化合物设5个浓度进入梯度复筛),每孔终体积200 μL,每种处理均设3个复孔.
3)显色:37oC培养48 h后,每孔加MTT溶液20 μL.继续孵育4 h,终止培养,吸弃孔内培养上清液,每孔加200 μL的SDS溶液(10%),过夜孵育(温度37oC),使结晶物充分融解.
4)比色:选择595 nm的波长,酶联免疫检测仪(Bio-Rad 680)读取各孔光吸收值,记录测定结果,以浓度为横坐标,细胞存活率为纵坐标绘制细胞生长曲线,应用两点法(Reed and Muench法)计算化合物的IC50值.
2 结果与讨论
3,4-羟基苯基丙烯酸甲酯在氧化银催化下,以无水甲苯和无水丙酮作为溶剂,得到苯并二氢呋喃环结构化合物1和苯并二氧六环类化合物2.该步反应与天然苯并二氢呋喃和苯并二氧六环类的生物合成途径类似[11],属自由基仿生氧化偶联反应,其反应机理如图2所示.Ag2O催化仿生氧化偶联法同时合成了两种新木脂素化合物,1和2的产量接近1∶1.该反应条件温和,后处理简单,以1/1.5倍当量新制氧化银粉末作催化剂最宜.经多次实验发现,采用未重蒸的甲苯和丙酮溶剂对产率并无太大影响,因此简化了实验条件.此外还发现,反应时间约40 h可达到较好收率,反应时间延长对产率提高不大且有可能增加其它副产物的生成.
化合物1用乙酸酐来保护酚羟基和DDQ脱氢反应,得到苯并呋喃类化合物4,化合物4的成功合成实现了苯并二氢呋喃向苯并呋喃环结构的转变,这为苯并呋喃新木脂素化合物的合成提供了一种有效的方法.在对酚羟基进行乙酰化保护的薄层色谱监测过程中,用稀盐酸先将反应液进行酸化,再点板,目的是消除溶剂吡啶在点板观察时的影响,此步反应可提高下一步氧化时的产率.实验发现,3.5倍当量的DDQ(2,3-二氯-5,6-二氰基-1,4-苯醌)可将原料全部脱氢氧化,后处理时以往采用的是硅胶柱过滤再经硅胶柱层析分离,虽然能达到尽可能除去DDQ的效果,但此过程需要使用大量的CH2Cl2且操作麻烦.因此,先用V丙酮/V甲醇=2∶1进行重结晶,再经硅胶柱层析分离得到纯品.
化合物4在经催化氢化得5,5在氢化铝锂作用和无水无氧操作条件下,室温进行还原反应得到含有二个醇羟基的苯并呋喃新木脂素6,同时还生成了部分还原的产物7.在用氢化铝锂还原时,一定要采用重蒸THF作溶剂,将溶有原料的THF溶液在-20oC的低温条件下缓慢滴加至氢化铝锂的THF溶液中,后处理用水猝灭反应时有大量氢气放出,所以加水过程一定要缓慢.化合物2在氢化铝锂作用和无水无氧操作条件下,室温进行还原反应则得到生物活性苯并二氧六环类天然产物isoamericanol A 化合物2经催化氢化和氢化铝锂还原分别得到未见文献报道的苯并二氧六环类化合物9和10.
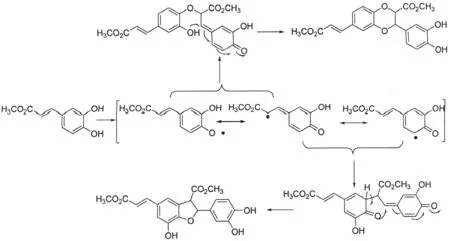
图2 苯并二氢呋喃和苯并二氧六环新木脂素类化合物的合成机理
对所合成的苯并呋喃新木脂素类化合物1,3~5使用MTT法进行生物活性的测试.半数生长抑制浓度IC50值表明,化合物1,3,4对白血病细胞(HL-60)、肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、结肠癌细胞(SW-480)、肝癌细胞(SMMC-7721)有明显的体外肿瘤生长抑制活性;化合物5对白血病细胞(HL-60)、肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、肝癌细胞(SMMC-7721)有明显的体外肿瘤生长抑制活性,其中部分抗肿瘤细胞活性优于对照药物顺铂(MW300)(如表1).
从1,3,4,5化合物的生物活性变化来看,羟基乙酰化的结构修饰明显提高了化合物对结肠癌细胞(SW-480)的抑制作用;由苯并二氢呋喃变为苯并呋喃的结构变化提高了化合物对白血病细胞(HL-60)和结肠癌细胞(SW-480)的生长抑制活性;8位、9位的乙烯基还原为乙基的结构变化使得化合物5对肺癌细胞(A-549)、乳腺癌细胞(MCF-7)、结肠癌细胞(SW-480)和肝癌细胞(SMMC-7721)的生长抑制活性降低,但其对白血病细胞(HL-60)的抑制活性优于化合物3.

表1 化合物对不同肿瘤细胞株的半数生长抑制浓度IC50
[1]CHAILIN KAO,JIWAN CHEM.A novel strategy for the synthesis of benzofuran skeleton neolignans,:application to ailanthoidol,XH-14 and obovaten [J].J Org Chem,2002,67:6772-6787.
[2]蒲文臣,王飞,王淳.2-芳基苯并[b]呋喃衍生物的生物活性与合成策略[J].有机化学,2011,31:155-165.
PU Wen-chen,WANG Fei,WANG Chun,Bioactivities and synthetic methods of 2-arylbenzo[b]furans[J].Chin J Org Chem,2011,31:155-165.(In Chinese)
[3]RAKOTONDRAMANANA D L,DELOMENEDE M,BALTAS M,etal.Synthesis of ferulic ester dimers,functionalisation and biological evaluation as potential antiatherogenic and antiplasmodial agents[J].Bioorg Med Chem,2007,15(18):6018-6026.
[4]FAN Hua-fang,REN Ying-mei,WU Xiu-ling,etal.Synthesis and cytotoxicity of novel benzofuran neolignan derivatives[J].J Chem Res,2010,34(4):233-235.
[5]WU Zheng,LIANG Zhi-ying,LI We,etal.Synthesis of (+)-Demethylnitidanin,Herpetol and Salvinal as well as their glycosyl derivatives[J].Chem Res Chinese Univertsities,2011,27(6):949-954.
[6]PIETERS L,VAN DYCK S,GAO M,etal.Synthesis and biological evaluation of dihydrobenzofuran lignans and related compounds as potential antitumor agents that inhibit tubulin polymerization[J].Journal of Medical Chemistry,1999,42(26):5475-5481.
[7]HITOSHI T,ICHIRO K.Total synthesis of cleomiscosin A,a courmari-neolignoid[J].Chem Pharm Bull,1985,33:3218-3223.
[8]FUKUYAMA Y,HASEGAWA T,TODA M,etal.Structure of americanin A and isoamericanol A having a neuotrophic property from the sead ofphytolaccaamericana[J].Chem Pharm Bull,1992,40(1):252-254.
[9]WANG E C,WEIN Y S,KUO Y H.A concise and effcient synthesis of salvinal from isoeugenolviaa phenoxenium ion intermediate[J].Tetrahedron Lett,2006,47(52):9195-9197.
[10]余竟光,李彤梅,孙兰,等.鹰爪种子化学成分的研究[J].药学学报,2001,36(4),281-286.
YU Jing-guang,LI Tong-mei,SUN Lan,etal.Studies on the chemical constituents of the seeds fromartabostryshexapeta(Annonaceae)[J].Acta Pharm Sin,2001,36(4):281-286.(In Chinese)
[11]KUO Y H,WU C H.Synthesis of 5-(3-Hydroxypropyl)-7-methoxy- 2-(3-methoxy-4-hydroxyphenyl)-3-benzo[b]furancarbaldehyde,a novel adenosine A1 receptor ligand from the root ofSalviamiltiorrhiza[J].J Nat Prod,1996,59(6):625-628.
猜你喜欢
中山大学学报(自然科学版)(中英文)(2021年3期)2021-05-26
中国粮油学报(2019年4期)2019-07-12
中成药(2018年2期)2018-05-09
湖北农业科学(2017年24期)2018-01-27
上海农业科技(2016年6期)2016-12-23
新乡学院学报(2016年6期)2016-12-01
环境科技(2016年4期)2016-11-08
中成药(2016年8期)2016-05-17
当代化工研究(2016年9期)2016-03-20
中国兽药杂志(2014年7期)2014-11-23
