
红景天苷的全合成*
2014-08-30 09:51张翼轩张昌浩赵春晖付晓磊陈艳华赵龙铉
合成化学 2014年6期
张翼轩,张昌浩,赵春晖,付晓磊,陈艳华,李 常,李 镐,赵龙铉,
(1.辽宁师范大学 a.化学化工学院;b.生物技术与分子药物研发辽宁省重点实验室,辽宁 大连 116029;2.延边大学 药学院 长白山生物资源与功能分子教育部重点实验室,吉林 延吉 133002)
·研究简报·
红景天苷的全合成*
张翼轩1a,张昌浩2,赵春晖1b,付晓磊1a,陈艳华1a,李 常1a,李 镐2,赵龙铉1a,1b
(1.辽宁师范大学 a.化学化工学院;b.生物技术与分子药物研发辽宁省重点实验室,辽宁 大连 116029;2.延边大学 药学院 长白山生物资源与功能分子教育部重点实验室,吉林 延吉 133002)
以对羟基苯甲醛为原料,经酚羟基保护、醛基还原、氯代、氰化、水解、酯化和还原反应制得关键中间体4-苄氧基苯乙醇(6);6与溴代四乙酰基葡萄糖偶联后脱保护基合成了红景天苷,总收率12.8%,其结构经1H NMR,13C NMR,IR和HR-MS确证。
红景天苷;4-苄氧基苯乙醇;全合成
红景天苷(8)是红景天最主要的有效成分之一,药用价值较高。研究发现,8除了具有抗辐射作用,可以作为癌症患者放疗前的保护用药之外[1-3],还具有抗缺氧、抗衰老、抗心肌缺血、抗癌、抗菌和提高免疫力等作用[4-7]。因此,8在中药和保健品行业具有广阔的市场前景。
目前,合成8常见的方法有以下几种:(1)Endo等[8]以对羟基苯乙醇和溴代四乙酰基葡萄糖(Ⅰ)为原料,经偶联反应合成8;(2)纪淑芳等[9]以对氨基苯乙醇为原料,经重氮化、水解等多步反应合成8,但反应条件较为复杂;(3)李国青等[10-11],以对羟基苯乙酸乙酯为原料,经苄基化、还原、成苷、脱保护基等步骤合成8,该方法原料价格相对较贵;(4)张三奇等[12],以对溴苯酚为原料,经烯丙基化、环氧化、苷化和脱烯丙基等多步反应合成8,此法反应条件苛刻,不易控制,且对实验设备要求较高。
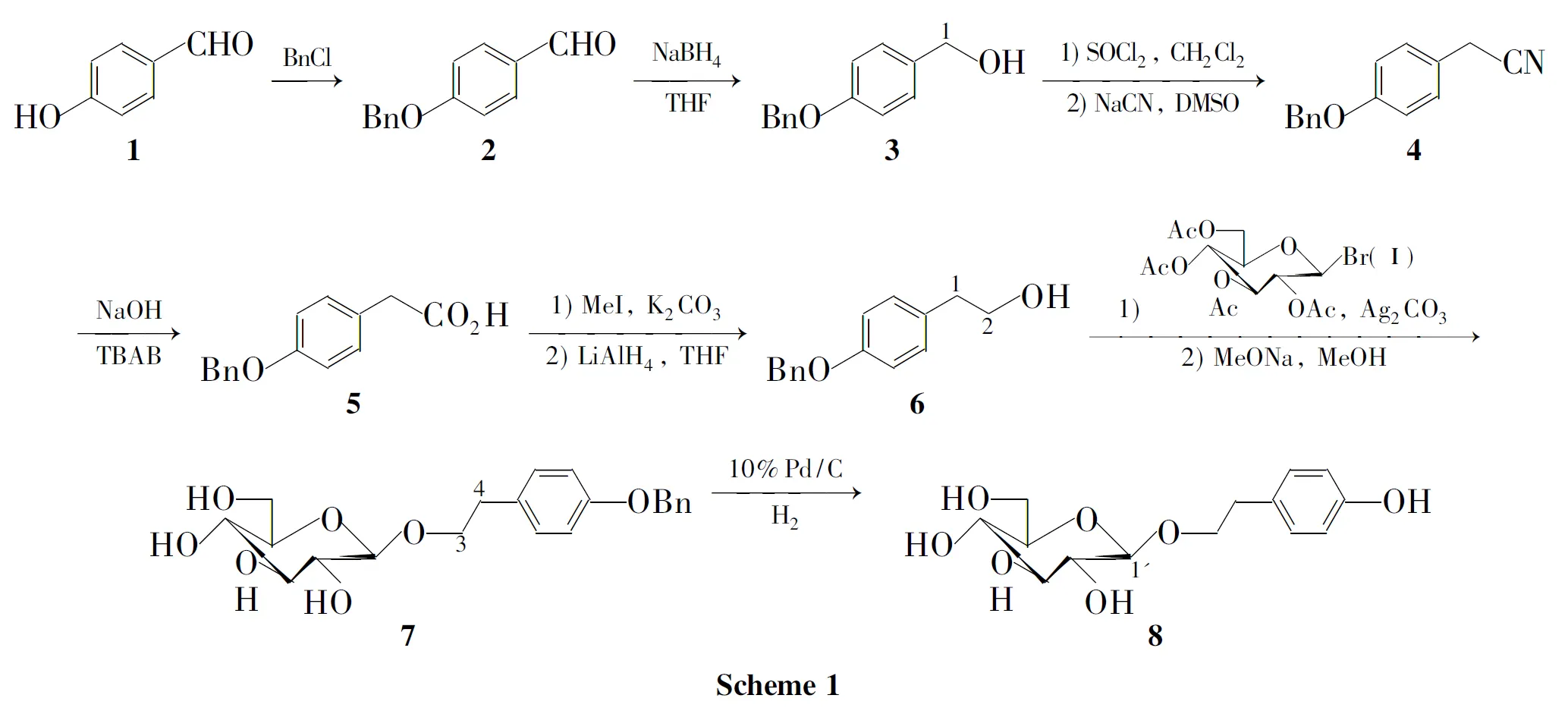
为降低成本,提高收率,寻求一种条件温和且易于控制的合成8的方法具有重要的意义。本文以结构更为简单的4-羟基苯甲醛(1)为原料,经酚羟基保护、醛基还原、氯代、氰化、水解、酯化和还原反应制得关键中间体4-苄氧基苯乙醇(6);6与Ⅰ偶联后脱保护基合成了8,总收率12.8%,其结构经1H NMR,13C NMR,IR和HR-MS确证。
1 实验部分
1.1 仪器与试剂
X-5型显微熔点仪(温度未校正);Bruker 500MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);WGH-30型双光束红外分光光度计(KBr压片);GC-Tof MS型高分辨率质谱仪。
Ⅰ按文献[13-14]方法合成;其余所用试剂均为分析纯。
1.2 合成
(1)4-苄氧基苯甲醛(2)的合成
在反应瓶中加入乙醇20mL和水10mL,搅拌下加入氢氧化钠1mg(29.47mmol),于75℃加入13.0g(24.56mmol),搅拌10min;滴加氯苄3.4mL(29.47mmol),滴毕,反应3h。减压蒸除乙醇,加水100mL,用二氯甲烷(3×30mL)萃取,合并有机相,用无水硫酸镁干燥,浓缩后经硅胶柱层析[洗脱剂:A=V(乙酸乙酯)∶V(石油醚)=1∶5]纯化得白色固体24.96g,收率95.2%,m.p.72.4℃~73.6℃;1H NMRδ: 9.91(s,1H,CHO),7.87~7.08(m,9H,ArH),5.17(s,2H,CH2);IRν: 3067,2850,2740,1690,1592,1575,1450,830cm-1。
(2)4-苄氧基苯甲醇(3)的合成
在反应瓶中加入硼氢化钠1.0g(26.88mmol),干燥的THF 30mL,搅拌下加入24.8g(22.42mmol),于室温反应3h(TLC监测)。滴加1mol·L-1盐酸调至无气泡产生为止,加水100mL,用二氯甲烷(3×30mL)萃取,合并有机相,用无水硫酸镁干燥,浓缩后经硅胶柱层析(洗脱剂:A)纯化得白色固体34.5g,收率93.3%,m.p.84.1℃~85.3℃;1H NMRδ: 7.39~6.97(m,9H,ArH),5.09(s,2H,CH2in Bn),4.63(s,2H,1-H);IRν: 3361,3048,2912,2858,1606,1579,1442,1225cm-1。
(3)4-苄氧基苯乙腈(4)的合成
在反应瓶中加入二氯亚砜7.5mL(102.98mmol)和二氯甲烷10.6mL(164.72mmol),冰浴冷却,搅拌下缓慢滴加34.4g(20.59mmol)的二氯甲烷(25mL)溶液,反应4h。滴加10%碳酸氢钠溶液至无气泡产生为止,加水100mL,用二氯甲烷(3×30mL)萃取,合并有机相,用10%碳酸氢钠洗涤,无水硫酸镁干燥,浓缩得A。
在反应瓶中加入氰化钠1.1g(22.94mmol)和二甲基亚砜15mL,充分搅拌下滴加A 3.5g(14.82mmol)的二甲亚砜(20mL)溶液,滴毕,于25℃反应2.5h。加入饱和食盐水50mL,用乙酸乙酯(3×30mL)萃取,合并有机相,用无水硫酸镁干燥,浓缩后经硅胶柱层析(洗脱剂:A)纯化得白色固体43.0g,收率84.5%,m.p.62.7℃~63.5℃;1H NMRδ: 7.45~6.98(m,9H,ArH),5.08(s,2H,CH2in Bn),3.69(s,2H,1-H);IRν: 3042,2919,2853,2239,1600,1584cm-1。
(4)4-苄氧基苯乙酸(5)的合成
在反应瓶中依次加入43.4g(15.03mmol),正四丁基溴化铵(TBAB)67mg,氢氧化钠1.9g(47.19mmol),水1.3mL和二甲苯3.8mL,搅拌使其溶解;于120℃反应6h。降温至5℃~10℃,用浓盐酸调至pH 1,反应3h。加水100mL,用乙酸乙酯(3×30mL)萃取,合并有机相,用无水硫酸镁干燥,浓缩后经硅胶柱层析(洗脱剂:A)纯化得白色固体53.2g,收率89.0%,m.p.110.8℃~111.5℃;1H NMRδ: 7.44~6.95(m,9H,ArH),5.07(s,2H,CH2in Bn),3.61(s,2H,1-H);IRν: 3108,1614,1457,1408,1121,1074,861,779cm-1。
(5)4-苄氧基苯乙醇(6)的合成
在反应瓶中加入52.5g(10.17mmol),碳酸钾3.9g(28.47mmol)和DMF 15mL,搅拌使其溶解;滴加碘甲烷1.4mL(22.37mmol),滴毕,于室温反应6h。加入饱和食盐水50mL,用乙酸乙酯(3×30mL)萃取,合并有机相,用无水硫酸镁干燥,浓缩后经硅胶柱层析(洗脱剂:A=1∶10)纯化得白色固体4-苄氧基苯乙酸甲酯。
在反应瓶中加入氢化铝锂268mg(7.08mmol),冰浴冷却,加入干燥THF 10mL,充分搅拌使其变为乳浊液,缓慢滴加4-苄氧基苯乙酸甲酯1.2g(4.72mmol)的THF(15mL)溶液,滴毕,回流(85℃)反应1h。用硅藻土抽滤,滤液浓缩后经硅胶柱层析(洗脱剂:A)纯化得白色固体60.940g,收率63.3%,m.p.79.1℃~80.4℃;1H NMRδ: 7.46~6.94(m,9H,ArH),5.07(s,2H,CH2in Bn),3.84(t,J=6.6Hz,2H,2-H),2.83(t,J=6.6Hz,2H,1-H);13C NMR(125MHz)δ: 157.56,137.14,130.76,129.99,128.58,127.93,127.44,115.06,70.10,63.79,38.31;IRν: 3180,3040,1607,1519,1463,1124,1059cm-1;HR-MSm/z:Calcd for C15H16O2[M]228.2863,found 228.1150。
(6)4-苄氧基苯乙基-β-D-葡萄糖苷(7)的合成
在反应瓶中加入6286mg(1.25mmol)和干燥二氯甲烷10mL,充分搅拌下加入碳酸银517mg(1.87mmol),分子筛(350℃活化)715mg和Ⅰ 771mg(1.87mmol),于室温避光反应36h。抽滤,滤饼用适量二氯甲烷洗涤,合并滤液和洗液,依次用饱和碳酸氢钠(30mL)和饱和氯化钠(3×20mL)洗涤,无水硫酸镁干燥,浓缩后经硅胶柱层析(洗脱剂:A=1∶10)纯化得白色固体4-苄氧基苯乙基-β-D-四乙酰基葡萄糖苷。
在反应器中加入4-苄氧基苯乙基-β-D-四乙酰基葡萄糖苷191mg(0.34mmol)和无水甲醇8mL,搅拌使其溶解;加入甲醇钠277mg(5.13mmol),于室温反应4h。用醋酸调至pH 6,减压蒸除溶剂,浓缩后经硅胶柱层析[洗脱剂:B=V(甲醇)∶V(二氯甲烷)=1∶10]纯化得白色固体795mg,收率49.7%,m.p.104.6℃~105.7℃;1H NMR(CD3OD)δ: 7.35~6.88(m,9H,ArH),5.04(s,2H,CH2in Bn),4.29(d,J=7.8Hz,1H,CH),4.07~3.16(m,8H,2′,3′,4′,5′,6′-H),2.88(dt,J=2.1Hz,7.5Hz,2H,3,4-H);IRν: 3445,3348,3267,2945,1615,1502,1454,1269,1020cm-1。
(7)8的合成
在反应瓶中加入7183mg(0.46mmol)和无水乙醇10mL,搅拌使其溶解;加入10%钯碳27mg,搅拌下于室温反应24h(氢气氛围)。过滤,滤液浓缩后经硅胶柱层析(洗脱剂:B)纯化得白色固体883.8mg,收率60.7%,m.p.156.7℃~158.4℃;1H NMRδ: 7.07~6.68(m,4H,ArH),4.30(d,J=7.8Hz,1H,CH),4.05~3.16(m,8H,2′,3′,4′,5′,6′-H),2.85(dt,J=2.8Hz,7.8Hz,2H,3,4-H);13C NMRδ: 156.77,130.91,130.77,116.12,104.36,78.10,77.93,75.11,72.06,71.66,62.77,36.35;IRν: 3283,2920,1607,1532,1443,1251,1026cm-1;HR-MSm/z:Calcd for C14H20O7[M]300.3044,found 300.1191。
2 结果与讨论
对酚羟基的保护主要有乙酰基保护和苄基保护[15-16]。实验结果显示,采用乙酰基作保护基,合成6和7的反应不易控制且产率较低。采用苄基保护,操作简便、容易纯化且产率相对较高。此外,也可利用此方法通过改变苯环上的取代基种类及位置合成7的类似物。
3 结论
本文以1为原料,经7步反应合成8,总收率12.8%。该方法具有原料易得,反应条件温和,总收率较高等优点,为8的全合成提供了一条新的合成路线。
[1] 刘三强,张棋,苑文房.红景天对大鼠辐射损伤防护作用的实验研究[J].临床医学工程,2010,17(2):50-51.
[2] 姜义,肖雪媛,关桂梅,等.红景天素对X射线照射小鼠的预防作用[J].中华放射医学与防护杂志,1995,15(3):214.
[3] 郑志清,叶于薇,董妙珠,等.红景天抗辐射功能的初步实验研究[J].上海预防医学杂志,2000,12(2):69-70.
[4] 苗艳波,师海波,孙英莲,等.高山红景天的抗辐射作用[J].中药药理与临床,2004,20(3):21-22.
[5] 叶子聪,陈钦铭,金凯平.红景天苷对培养心肌细胞缺氧后再给氧损伤后的影响[J].中国药理学报,1993,14(5):424-426.
[6] 张淑芹,孙非,刘志屹,等.高山红景天甙抑制白血病细胞生长的实验研究[J].吉林中医药,1999,4:56.
[7] Agnieska M,Hoser G,Furmanwa M,etal.Antiproliferative and antimitotic effectS-phase accumulation and induction of apoptosis and necrosis after treatment of extract from rhodiola rosea rhizomes on HL-60Cells[J].J Ethnopharmacol,2006,103(1):43-52.
[8] Endo K,Seya K,Hikino H.Biogenesis-like transformation of salidroside to rengyol and its related cyclohexyletanoids of forsythia suspens[J].Tetrahedron,1989,45(12):3673-3682.
[9] 纪淑芳,周亚青.红景天苷的合成[J].沈阳药学院学报,1987,4(3):192-194.
[10] 李国青,李 展.红景天苷合成方法的改进[J].中国药物化学杂志,1996,6(2):136-137.
[11] 张莲姬,李雪梅,田官荣.红景天苷的合成[J].延边大学学报,2002,28(2):97-98.
[12] 张三奇,尚刚伟,李中军,等.合成红景天苷的新途径[J].中国药物化学杂志,1997,7(4):256-257.
[13] 李玉文,李英霞,张伟,等.一锅法制备全乙酰吡喃溴代糖[J].有机化学,2004,24(4):438-439.
[14] Randazzo G,Capasso R,Rosaria M,etal.A simple method for detritylation of carbohydrate derivatives[J].Carbohyd Res,1980,85(2):298-301.
[15] Prichard W W.Hydroquinone diacetate[J].Org Synth,1995,3:452-453.
[16] Ma C,Nakamura N,Miyashiro H,etal.Inhibitory effects of constituents from cynomorium songaricum and related triterpene derivatives on HIV-1protease[J].Chem Pharm Bull,1999,47(2):141-145.
TotalSynthesisofSalidroside
ZHANG Yi-xuan1a,ZHANG Chang-hao2,ZHAO Chun-hui1b, FU Xiao-lei1a,CHEN Yan-hua1a,LI Chang1a,LI Gao2,ZHAO Long-xuan1a,1b
( a.Chemistry and Chemical Engineering;b.Liaoning Provincial Key Laboratory of Biotechnology and Drug Discovery,1.Liaoning Normal University,Dalian 116029,China;2.Key Laboratory of Natural Resources of Changbai Mountain and Functional Molecules,Ministry of Education,College of Pharmacy,Yanbian University,Yanji 133002,China)
A key intermediate,4-(benzyloxy)phenethyl alcohol(6),was prepared by the reaction of protection,reduction,chlorination,cyanidation,hydrolysis,esterification,reduction from 4-hydroxybenzaldehyde.Salidroside with total yield of 12.8% was synthesized by coupling reaction of6with tetra-O-acetyl-D-glucopyranosyl bromide,and then removed the acetyl group and the benzyl group.The structures were confirmed by1H NMR,13C NMR,IR and HR-MS.
Salidroside;4-(benzyloxy)phenethyl alcohol;total synthesis
2014-05-23
国家自然科学基金资助项目(81160386)
张翼轩(1987-),男,汉族,内蒙古赤峰人,硕士研究生,主要从事药物合成的研究。
赵龙铉,教授,硕士生导师,E-mal: lxzhao@lnnu.edu;李镐,教授,博士生导师,E-mal: gli@ybu.edu.cn
O625.31;R284.3
A
1005-1511(2014)06-0813-04
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
云南化工(2020年4期)2020-05-19
科海故事博览·中旬刊(2020年3期)2020-03-15
吉林农业(2019年6期)2019-06-11
安徽化工(2018年5期)2018-10-23
教育教学论坛(2018年38期)2018-09-25
铜仁学院学报(2018年6期)2018-07-05
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
中国洗涤用品工业(2016年2期)2016-02-28
