
孤独症谱系障碍致病基因SHANK3的研究进展
2014-08-11 14:47刘春雪姜永辉
中国循证儿科杂志 2014年4期
刘春雪 姜永辉 徐 秀
·综述·
孤独症谱系障碍致病基因SHANK3的研究进展
刘春雪1姜永辉2徐 秀1
孤独症谱系障碍(ASD)是一类以不同程度的社会交往/交流障碍、狭隘的兴趣和重复刻板行为、感知觉异常为主要特征的发育行为障碍性疾病,严重影响患者及其家庭的生活质量。
尽管目前ASD的病因在多数病例中仍不完全明了,但多数学者认为遗传因素、环境因素在ASD的发病中有重要作用。双生子研究显示,ASD同卵双生共患率达60%~92%,异卵双生共患率约10%,同胞再患概率3%~5%,较普通人群高25~60倍[1,2],说明ASD与遗传因素密切相关。近年来,采用候选基因及全基因组关联研究,发现多个与ASD有关的突触结构及功能相关的致病候选基因,如SHANK3、NLGN3、NLGN4x、CNTNAP2、NRXN1、NRXN2、PCD9等。
另外,DNA拷贝数变异(CNV)可改变基因剂量,导致不同程度的基因表达差异,对表型改变及疾病的发生发展具有一定作用[3,4]。研究发现,ASD患者较正常人更常携带CNV[5]。已报道SHANK3、FOXP2、NRXN1等基因的CNV及16p11.2、15q11-q13与ASD显著相关[6~9]。本文将重点介绍SHANK3(SH3 and Multiple Ankyrin Repeat Domains 3)分子缺陷与ASD发生的关系。
1 SHANK3基因
2007年,研究人员对来自3个家庭的5名ASD儿童进行检测,发现了与ASD有关的新基因——SHANK3[10]。到目前为止,已经在超过1 000例ASD患者中识别出SHANK3基因的6类分子缺陷[11]:①细胞遗传学检测可见的22q13.3缺失(5~10 Mb)或环状22号染色体[12,13],②微缺失(0.1~4 Mb)[14,15],③微小扩增[16],④基因内断点易位[17],⑤微小基因内缺失(<100 kb)[18],⑥点突变[10,14]等。近来发现SHANK3基因CpG丰富的序列(又称“CpG”岛,CGI)的高度甲基化能够改变其蛋白的组织特异性表达[19,20]。
人类基因组共有3个SHANK基因(SHANK1,SHANK2,SHANK3),分别表达在大脑的不同区域。其中,SHANK3是SHANK家族中与ASD关系最密切的基因[20]。SHANK3基因位于22号染色体的13.3长臂(22q13.3),全长约58 kb,包含22个外显子(NM_033517),且富含GC序列(GC含量高达69%)。SHANK3基因编码由1 747个氨基酸组成的SHANK3蛋白,该蛋白主要表达在兴奋性神经元的突触后致密区(PSD)。SHANK3蛋白在人类纹状体中呈高表达,尤其在神经元的突触中。在小鼠中,SHANK3蛋白在丘脑、纹状体和小脑颗粒细胞中呈高表达[21],在心脏、脾脏、小肠和肾脏等也均有表达,即SHANK3基因几乎存在于各个系统中,这可以解释ASD患者全身系统的功能改变。
PSD是突触后膜细胞骨架纤维特化的区域,后膜的细胞骨架和定位与突触前膜末端的活性区域相对应。这种结构的功能是调节细胞黏附性、控制受体聚集和调节受体功能[22]。通过PSD,细胞表面受体可以与肌动蛋白细胞骨架连接。在突触中,突触前膜的神经元表面蛋白(neurexin)结合到突触后膜的神经配体蛋白(neuroligin)上,神经配体蛋白再绑定到SHANK3蛋白上[23],并在谷氨酸能神经元突触上形成复合物。这种蛋白复合物在维持突触正常功能和树突棘正常形态,以及调节神经兴奋-抑制平衡中均发挥关键作用[21]。
SHANK3有多个蛋白结构域[11]:ANK、SH3、PDZ(PSD-95/Discs large/ZO-1)、富含脯氨酸结构域(proline rich region containing homer and cortactin-binding sites)和SAM(图1)。作为突触后致密复合体的主要支架蛋白,SHANK3蛋白与多种突触分子相互作用[11],可通过ANK结构域与细胞骨架结合,通过PSD-95/GKAP与膜上的NMDA受体或细胞黏附分子等结合,通过homer-binding结构域与mGlu受体结合等[20]。
研究显示,SHANK3基因在人工培养的海马神经元中过表达时,可促进树突棘的扩大和成熟,而敲除海马神经元的SHANK3基因时,树突棘的密度则减少[24]。这说明SHANK3能促进树突棘的成熟和扩大,甚至能够诱导无棘神经元的树突棘形成[25]。同样,SHANK3基因敲除小鼠表现出自残行为、重复理毛、社会交往和沟通障碍[26~28],其海马细胞神经传递减少、长时程增强作用异常、突触减少、树突分叉增多[29]、树突棘数量明显减少和PSD更稀薄等[30]。同时,一些支架蛋白,如鸟苷酸激酶相关蛋白(GKAP)和谷氨酸受体亚基的水平也降低,这与神经信号强度降低的结果一致[29,31]。这些研究结果提示,SHANK3基因在树突棘的形成、成熟和稳定中都起关键作用[32,33]。另外,尽管SHANK3基因敲除小鼠整个大脑体积无明显增加,但纹状体体积有少量增加,而在ASD患者中发现尾状核体积有明显增加。这些神经生物学的改变可能与ASD样行为有关,包括焦虑、刻板行为、自残行为和拒绝社会交往等[26,29]。

图1 人类SHANK3基因外显子结构、选择性剪接的外显子、蛋白结构域、启动子和5个CpG岛(CGI)[19]
注 11a是一个新发现的外显子;蓝色箭头表示目前在ASD患者中发现的可能致病的点突变位点
2 22q13.3缺失综合征
SHANK3基因最初被认为与22q13.3缺失综合征有关[34]。22q13.3缺失综合征,又称Phelan-McDermid综合征(PMS,MIM #606232),是由22号染色体长臂末端的微缺失所致[21]。首例PMS由Watt在1985年报道,1例4岁男孩患有严重智力障碍、无语言交流和面部畸形。研究发现,其缺失来源于母亲的22号染色体的臂间倒位,造成减数分裂重组,从而导致22q12至22q末端(22qter)的缺失。PMS的主要特征为全面发育迟缓、肌张力减退、中至重度智力障碍、语言缺失或严重的语言发育迟缓和颅面部异常[11,23]等,近来又发现PMS患者有非典型双向情感障碍[35,36]。在超过1 000例PMS患者中有75%以上被诊断为ASD[11],具有典型的ASD临床表现,如社会交往/社会沟通障碍、眼神交流减少、焦虑和自伤行为,因此PMS被列为ASD的一种综合征形式[23]。大部分22q13.3的末端缺失是由于简单的缺失、非平衡易位、环状染色体所致,缺失长度为100 kb至>9 Mb[23]。
Bonaglia等[34]发现1例男童具PMS全部特征,其神经系统损害尤为突出,表现为轻度智力低下、重度语言发育迟缓、轻度肌张力减退和典型的面部异常。遗传分析发现染色体存在平衡易位:46,XY,t(12;22)(q24.1;q13.3)。通过测序,在22号染色体位置296489-296494处发现断裂点。该研究首次证明了SHANK3是PMS患者神经损害(语言发育迟缓和智力障碍)的主效基因。另一方面,对微小末端缺失的研究进一步支持了SHANK3基因在PMS中的重要作用。Misceo等[37]在1例女性患者中也发现了SHANK3基因末端缺失,包括SHANK3基因最后2个外显子以及ACR基因,SHANK3最后2个外显子包含SAM结构域,提示SAM结构域丢失会引起PMS临床表型。核型分析发现:46,X,t(X;22)(q21.3;q13.33),测序发现缺失长度为17 581 bp。同时,该患者X染色体上存在283 764 bp的扩增。该患者4岁时即表现出严重的语言发育迟缓和社交障碍,并表现出刻板行为。6岁时诊断为中度智力发育迟缓,20岁时出现高促性腺激素性腺功能低下症。
2003年,Luciani等[38]在33例PMS患者中发现,17例存在环状22号染色体,片段缺失从160 kb至9 Mb,且关键区域包含SHANK3、ACR和RABL2基因。其中74%的缺失来源于父亲的22号染色体。同年,Wilson等[13]报道了 56例PMS患者,其22q13.3区域缺失长度为130 kb至>9 Mb。同样,大部分PMS病例(45/56)缺失的关键区域也包含SHANK3基因,且父亲的22号染色体缺失占其研究病例的70%。这种高比例的父源缺失也在其他末端缺失综合征中发现[39,40]。这些现象表明,与女性生殖细胞相比,男性生殖细胞对染色体断裂可能具有更大的易感性。
22q13.3缺失综合征可能缺失的基因超过90种[23],其中神经系统障碍与SHANK3基因关系最为密切,主要是由SHANK3单倍剂量不足引起[23]。22q13.3区域的缺失造成此类PMS患者的每个细胞的SHANK3基因只有1个拷贝数,而不是正常的2个拷贝数。小鼠模型提示SHANK3单倍剂量不足造成了SHANK3蛋白的表达减少50%[41],引起突触功能和神经元之间信息传导功能的下降,从而导致PMS的临床特征。
Shcheglovitov等[42]从PMS患者的皮肤成纤维细胞中提取诱导性多功能干细胞(iPS cells),并进一步用来产生功能性神经元。研究显示,PMS神经元的SHANK3基因表达减少,并造成兴奋性突触传递出现严重缺陷,而对于抑制性突触传递则无明显影响。而这些PMS神经元的兴奋性突触传递可以通过SHANK3基因的再表达或者使用胰岛素样生长因子1(IGF1)获得纠正。
3 SHANK3基因扩增
研究表明,22号染色体长臂末端(22qter)无论是缺失还是扩增均与ASD有关,表现为神经功能障碍,SHANK3基因就位于此区域。22q近端段的扩增比较常见,而22q末端扩增则较为罕见。Barajas-Barajas等[43]总结了22q末端扩增患者的临床特征,包括严重智力障碍、生长发育迟缓、生长缺陷、先天性肌张力低下、脑水肿、小头畸形、内眦赘皮、耳位低、鼻梁突出、腭裂、人中长、小颌畸形和隐睾症等。
表1中总结了文献中报道的21例22q12或22q13-22qter的单纯扩增(不伴有其他染色体的缺失),其中13例由家族性平衡易位(9例是母方,4例是父方)引起。这些患者的平均寿命均较低。9/21例在12岁前夭折。这21例分为4组:22q13-22qter的扩增(8例)、22q13.1-22qter的扩增(2例)、22q13.2-22qter的扩增(3例)、22q13.3-22qter的扩增(8例)。其中22q13.1-22qter扩增病例存在多器官发育缺陷,临床症状突出,生存率也低于另外3组。而22q13.2-22qter和22q13.3-22qter的生存率较高,但其临床症状差异较大[44]。
Feenstra等[44]报道了首例成人男性22q13扩增患者,21号和22号染色体存在易位:46,XY,der(21)t(21;22)(p12;q13.3),其临床表型轻微,只有轻度的学习障碍和轻微的面部异常,其儿子也存在相同的易位,表现为面部畸形(眼睛深陷、面中部平坦、上唇突出),精神运动发育迟缓,惊厥发作,社会交往障碍 。 而该患者父母的基因型和表型都正常。
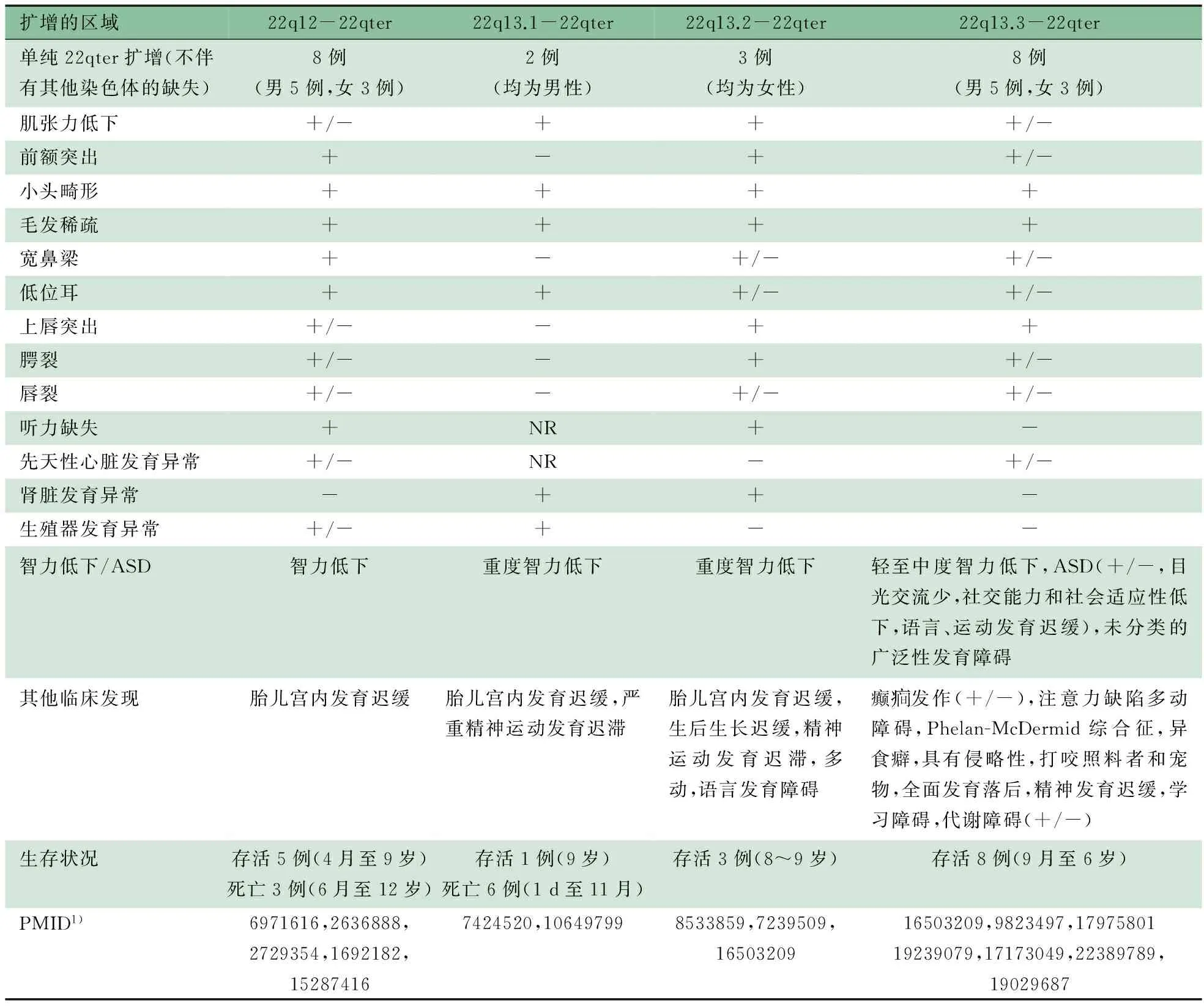
表1 4种22q末端单纯扩增及其临床表现
注 1)PMID为PubMed indexed唯一标识码。+:阳性,-:阴性;NR :未报道
Jafri等[45]首次报道了一个家庭中两兄弟同时存在22q13.3扩增和22q13.3缺失,其母亲22号染色体上存在倒位:46, XX, inv(22)(p13; q13.32)。该母亲的表型正常,曾流产过2次,现存活3个儿子。次子6岁,婴儿时期有轻度发育迟缓、社交能力和适应能力障碍,曾被诊断为未分类的广泛性发育障碍(PDD-NOS),基因检测发现其22号染色体上有22q13.3-22qter的扩增。三子4岁,婴儿时期有严重的全面发育落后,尤其是语言和运动发育迟缓,并伴有面部畸形特征:睑裂、肉质手、宽鼻梁、面中部平坦、下颌和耳朵突出,先天性趾侧弯,痛阈高,摇晃步态(肌张力低下),出现ASD样行为(具有攻击性,如打咬照料者和宠物),呈典型的PMS特征,曾被诊断为广泛性发育障碍和注意力缺陷多动障碍(ADHD)。基因检测发现其22号染色体上有22q13.3-22qter缺失。该家系提示当亲代存在倒位或平衡易位时,可能会以非平衡易位的形式传递给后代,或者通过正常的等位基因和倒位的等位基因之间发生重组而出现缺失和扩增。
22q末端扩增可能导致位于此区域的SHANK3基因过表达,干扰突触发育,如Okamoto等[16]报道的1例22q13微扩增的6岁女童,2岁4月才会独走,生长发育迟缓,6岁只能理解一些简单的句子,说几个单词,MRI显示额叶白质区域信号强度异常(T1低信号,T2高信号)。SHANK3单倍剂量不足已证实与ASD有关[10],但SHANK3扩增与ASD的关系还不明确。Durand等[10]发现1例22q末端出现扩增的男童,诊断为Asperger综合征,语言发育较早,语言流利,但社会沟通和人际交往表现出严重的障碍。类似的发现也在其他报道中出现[46]。但并不是所有的SHANK3基因过表达都引起ASD[16,47],而且在一项针对427例ASD患者进行的全基因组CNV分析结果中未发现22qter扩增[48]。因此仍需要进一步探究SHANK3基因扩增与神经系统异常有何种重要的联系以及其与ASD的关系。另外,22q13.3-22qter扩增的病例数可能比现有报道的更多,只是由于其片段太小以及临床症状相对较轻,目前的诊断技术尚未能全部诊断出来[48]。
4 SHANK3基因突变
目前,在约1%的ASD患者中检测到SHANK3基因突变[14]。在ASD患者中发现SHANK3基因的序列改变,包括错义突变、移码突变和剪接位点的突变[10, 14, 49~52]。图1中的蓝色倒三角显示人类中已发现的SHANK3点突变位置。表2总结了近5年来国内外发现的与ASD有关的SHANK3基因突变类型,以及突变体基因型/表型的关系。在ASD患者和严重的语言发育障碍患者中发现了新的点突变,其影响内含子5的剪接受体位点[31]和内含子9的剪接供体位点[50],而外显子21的一个碱基对插入突变则引起框移(p.A1227fs)[10]。p.A1227fs突变的患者表现为非典型性ASD和语言发育迟缓,此突变是从其父亲遗传,其父也有学习障碍和ADHD[14]。而剪接突变c.1820-4G>A与Asperger综合征患者有关[14]。以上数据表明,SHANK3分子缺陷能够引起ASD,但其临床表现差异较大。
大片段缺失一般与明显的结构畸形和社会交流障碍有关,而小片段缺失或点突变往往表现为与SHANK3单倍剂量不足有关,包括ASD、惊厥、异常EEG、肌张力减退、睡眠障碍、异常脑MRI和胃食管反流[53]。虽然在ASD患者中发现SHANK3基因缺失提示单倍剂量不足是ASD发病的主要原因[13],但对于SHANK3基因点突变(如错义突变和小的基因内缺失)的发病机制并不是很清楚[18,54]。另外,在ASD患者中,SHANK3基因致病性突变的频率不足0.75%[55],因而对ASD的贡献较小。不同的SHANK3基因突变可能会通过不同的机制来调节PSD中蛋白-蛋白相互作用,引起突触功能改变,从而造成各种不同的临床表型[11]。但是,结合本文中总结的基因型-表型的联系可以给临床诊断提供思路。
5 SHANK3基因的甲基化
SHANK3基因除了发生缺失、扩增和点突变外,其CGI的异常甲基化也与ASD的发病相关。组织特异性的DNA甲基化被认为是调节基因表达的重要方式,它不改变DNA的一级结构,却能改变基因的表达。DNA甲基化常发生在基因5′端的CGI。甲基化程度与基因表达通常呈反比关系,即CGI高度甲基化往往引起基因的低表达。
SHANK3基因是一个高GC含量的基因,有多个选择性剪接外显子(图1中红色标记),这些选择性剪接外显子分别编码SH3、proline-rich和 SAM结构域。SHANK3有5个CGI[56],图1显示,1个位于5′启动子区和4个位于基因内(基因内CGIs)。这些CGI在脑内表现为局部区域特异性的甲基化模式。Maunakea等[57]首次报道SHANK3基因中存在一个以上的基因启动子活性,Wang等[58]进一步证实小鼠的SHANK3基因中有6个基因启动子,这些基因内多个启动子以及选择性剪接使SHANK3基因产生多个mRNA和蛋白异构体,而每个SHANK3异构体均能与不同的蛋白结构域形成一种独特的组合方式[58]。
Beri等[27]分析了小鼠和人SHANK基因的CGIs的DNA甲基化,发现SHANK1、SHANK2和SHANK3基因均含有CGIs,但只有SHANK3基因在其大部分的CGIs中显示组织特异性的甲基化模式,而且当SHANK3基因在组织中呈高度甲基化时,其表达很低甚至缺如。研究显示,经体外甲基化处理后,海马神经元中SHANK3蛋白的表达显著减少。相反,HeLa细胞在经甲基化抑制剂5-氮杂-2'-脱氧胞苷(5-AZA)处理后,SHANK3的CGIs出现去甲基化,又重新表达SHANK3蛋白,SHANK3异构体的特异性表达也发生改变[32]。因此,减少或增加基因组DNA甲基化,均能调节SHANK3基因在蛋白质水平的表达。提示DNA甲基化的改变和脑SHANK3基因的表达、ASD患者的神经系统障碍之间存在密切的联系。

表2 人类SHANK3基因突变类型和表型的关系
注 1)PMID为PubMed indexed唯一标识码
Zhu等[19]分析54例ASD患者和43例对照组脑组织SHANK3基因的5个CGIs的DNA甲基化水平,在约15%的ASD患者脑组织中发现CGI-2、3和 4的所有CpG位点的甲基化水平都比对照组高,并发现特定SHANK3 mRNA异构体的表达水平的降低以及选择性剪接的改变等转录调节异常复杂的模式。亚硫酸氢盐克隆和测序研究(BSP)[27]的结果也相似,SHANK3基因的CGI-2~5在外周血淋巴细胞(PBL)中表现出广泛的甲基化,但在大脑、小脑和心脏中甲基化程度很低,甚至缺如。其中,CGI-2比较特殊,其所有的CpG二核苷酸在小脑以及大部分的大脑和心脏中未发生甲基化。而CGI-1在所有的组织中几乎都不发生甲基化,CGI-5的甲基化水平也较低。提示SHANK3基因内CGIs具有组织特异性的基因表达和选择性剪切作用[57,59]。
6 SHANK3突变小鼠模型
当前,对于以ASD患儿为样本的机制研究存在很多的局限性:①尸检脑组织资源非常有限,并且通常不能获得高质量的脑组织;②神经影像学研究提供的信息有限;③由于ASD患儿在临床症状和分子缺陷上存在较大的异质性,使研究设计和数据解释存在种种困难。因此,建立与人类ASD具有相同分子缺陷和临床表型的动物模型是研究ASD的关键。小鼠模型对于研究基因功能、探索SHANK3基因在ASD神经病理学中的作用具有重要意义。
突触后SHANK3蛋白在正常的神经连接发育中起关键性的作用,研究人员发现SHANK3基因敲除后的小鼠表现出ASD的特征,包括社会行为缺陷、刻板行为、自残行为和焦虑等,并出现异常的突触形态[30,58](如树突棘密度减少,长度增加)和异常的突触生理功能[28,30](如AMPA受体介导的突触传递减弱、微小兴奋性突触后电流频率增加和长时程增强作用减弱等)。这些发现表明,SHANK3基因在神经连接中起关键作用,并为以后探索其他社会行为的神经遗传学改变提供了一种新的思路[28]。通过神经元细胞培养[54,60]和小鼠模型[28,30,58,61]证明了SHANK3在纹状体水平调节PSD,在AMPA谷氨酸受体上调节基底神经传递,影响长时程增强作用,通过组织AMPA受体转运重塑树突棘等过程中起重要作用。
如今报道的SHANK3基因突变小鼠的突变类型主要包括编码ANK结构域的外显子4~9缺失[30,58]、编码ANK结构域的外显子4~7缺失[28]、编码SH3结构域的外显子11缺失[41]和编码PDZ结构域的外显子13~16缺失[28]。外显子4~9缺失小鼠GluA1、GKAP、Homer1b/c和GluN2A减少,表现为社会新奇感减少,二元测试中双向社会互动降低[30,58]。外显子4~7缺失小鼠皮质纹状体的突触传递轻度减少,但能正常发起社会交往[28]。外显子11缺失小鼠GluN2B和SHANK2增加,出现自残行为[41]。外显子13~16缺失小鼠SAPAP3/GKAP3、PSD-93、Homer1、GluN2A和GluN2B减少,纹状体体积、树突长度和表面积均增加,但树突棘密度以及长度减少,二元测试中互动减少,鼻-鼻接触频率降低[28]。

SHANK3基因在哺乳动物胚胎和新生儿早期阶段的脑中的表达相对较低,在出生后2周表达增强,成人大脑中又低于发育中的大脑[63,64]。小鼠模型提示CGI-2的甲基化率在出生后1周明显增高,高峰是2周,然后逐渐下降[20]。小鼠大脑的突触形成主要发生在生后2周内,随着突触的成熟,SHANK3的表达增加[64]。有趣的是,CGI-2在突触成熟的过程中甲基化模式发生变化。SHANK3基因的转录开始于CGI-2附近,其表达在出生后增强,但在第2周短暂减弱,之后又再次增强。
7 总结与展望
神经元之间准确无误的信息交换是通过突触实现的,突触传递活动是学习、记忆和认知的生物学基础。SHANK3基因编码的SHANK3蛋白是位于兴奋性PSD的多结构域支架蛋白,主要功能是将经递质受体,离子通道和其他膜蛋白与肌动蛋白细胞骨架和G-蛋白偶联的信号传导途径连接起来。与神经生长素共同参与了突触后结构的构建,尤其是那些与语言和社会交往相关的突触结构通路。另外,SHANK3蛋白在突触的形成和树突棘的成熟中具有重要作用。
异常的SHANK3基因剂量与严重的认知缺陷有关,包括语言和交流障碍以及ASD。SHANK3基因的单倍剂量不足与22q13.3缺失综合征有重要联系,而SHANK3基因过表达则可能表现出躁狂样的行为和惊厥发作。小脑颗粒细胞中的SHANK3基因过表达通过结合谷氨酸的不同亚型受体来减少树突棘和突触的形成,相反,抑制海马神经元SHANK3基因的表达则会减少树突棘的数量[27]。因此,任何一个方向上(表达不足或过度表达)的不正确基因剂量都可能是有害的。
尽管遗传因素在ASD发病机制中占有非常重要的作用,但基因突变和染色体微缺失或重复只占特发性ASD的10%~20%,大多数特发性ASD的病因依然不明[19,59,65,66]。越来越多的研究发现,基因和环境的相互作用对ASD的发生起关键作用[67]。在ASD患儿中,可能并不一定存在DNA水平的变异,而是基因的调控水平发生异常,某些目前未知的环境因素起着重要作用[68]。脑内突触功能障碍会引发ASD[69],进而可以推测突触基因(尤其是SHANK3基因)的表观遗传失调(如DNA甲基化)可能是ASD的分子基础。CGIs已经在人类50%的基因中发现,且通常位于启动子或外显子区域[27]。SHANK3基因的DNA甲基化从分子的层面提示,CGIs与SHANK3的异构体和组织特异性的表达有关,SHANK3基因在ASD的发病机制中可以作为基因与环境相互作用的一个有效的生物学标志。
结合生理学、神经病理学和行为学的研究,小鼠模型对于研究基因功能、探索SHANK3基因在ASD发生中具有关键作用。另外,SHANK3为相对保守的基因,斑马鱼与人的序列同源性核苷酸为61.9%,氨基酸为65.5%。斑马鱼作为模式动物,不仅可以通过检测分子基因水平变化,检测细胞组织器官发育分化及形态差异,还可通过分析其行为特征,对神经精神系统的发育进行研究。因此,可以利用动物模型来探讨SHANK3基因分子缺陷在ASD发生形态、行为变化中的分子机制,为ASD的分子靶向干预提供新的思路。
[1]Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry, 2011,68(11):1095-1102
[2]Kuehn BM. CDC: autism spectrum disorders common. JAMA, 2007,297(9):940
[3]Constantino JN, Zhang Y, Frazier T, et al. Sibling recurrence and the genetic epidemiology of autism. Am J Psychiatry, 2010,167(11):1349-1356
[4]Larsson HJ, Eaton WW, Madsen K M, et al. Risk factors for autism: perinatal factors, parental psychiatric history, and socioeconomic status. Am J Epidemiol, 2005,161(10):916-925, 926-928
[5]Tan Q(谭琪), Zeng YF. 遗传变异的又一来源:拷贝数变异. Letters in Biotechnology(生物技术通讯), 2009, 20(3):396-398
[6]Gong X, Jiang YW, Zhang X, et al. High proportion of 22q13 deletions and SHANK3 mutations in Chinese patients with intellectual disability. PLoS One, 2012,7(4):e34739
[7]Gauthier J, Champagne N, Lafreniere RG, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A, 2010,107(17):7863-7868
[8]Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med, 2008,358(7):667-675
[9]Bucan M, Abrahams BS, Wang K, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet, 2009,5(6):e1000536
[10]Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet, 2007,39(1):25-27
[11]Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron, 2013,78(1):8-27
[12]Jeffries AR, Curran S, Elmslie F, et al. Molecular and phenotypic characterization of ring chromosome 22. Am J Med Genet A, 2005,137(2):139-147
[13]Wilson HL, Wong AC, Shaw SR, et al. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet, 2003,40(8):575-584
[14]Boccuto L, Lauri M, Sarasua SM, et al. Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet, 2013,21(3):310-316
[15]Dhar SU, Del GD, German JR, et al. 22q13.3 deletion syndrome: clinical and molecular analysis using array CGH. Am J Med Genet A, 2010,152A(3):573-581
[16]Okamoto N, Kubota T, Nakamura Y, et al. 22q13 Microduplication in two patients with common clinical manifestations: a recognizable syndrome?. Am J Med Genet A, 2007,143A(23):2804-2809
[17]Bonaglia MC, Giorda R, Mani E, et al. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet, 2006,43(10):822-828
[18]Bonaglia MC, Giorda R, Beri S, et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with Phelan/McDermid syndrome. PLoS Genet, 2011,7(7):e1002173
[19]Zhu L, Wang X, Li XL, et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum Mol Genet, 2014,23(6):1563-1578
[20]Uchino S, Waga C. SHANK3 as an autism spectrum disorder-associated gene. Brain Dev, 2013,35(2):106-110
[21]Guilmatre A, Huguet G, Delorme R, et al. The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol, 2014,74(2):113-122
[22]Sheng M. Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci U S A, 2001,98(13):7058-7061
[23]Phelan K, McDermid HE. The 22q13.3 Deletion Syndrome (Phelan-McDermid Syndrome). Mol Syndromol, 2012,2(3-5):186-201
[24]余金丹. 突触相关基因 NRXN1, NLGN3, NLGN4X, CNTNAP2, SHANK3 遗传变异与汉族儿童孤独症的相关性研究. 浙江大学, 2011
[25]Boeckers TM, Bockmann J, Kreutz MR, et al. ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem, 2002,81(5):903-910
[26]Yang M, Bozdagi O, Scattoni ML, et al. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J Neurosci, 2012,32(19):6525-6541
[27]Beri S, Tonna N, Menozzi G, et al. DNA methylation regulates tissue-specific expression of Shank3. J Neurochem, 2007,101(5):1380-1391
[28]Peca J, Feliciano C, Ting JT, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature, 2011,472(7344):437-442
[29]Herbert MR. SHANK3, the synapse, and autism. N Engl J Med, 2011,365(2):173-175
[30]Bozdagi O, Sakurai T, Papapetrou D, et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism, 2010,1(1):15
[31]Hamdan FF, Gauthier J, Araki Y, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet, 2011,88(3):306-316
[32]Sala C, Piech V, Wilson NR, et al. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron, 2001,31(1):115-130
[33]Roussignol G, Ango F, Romorini S, et al. Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci, 2005,25(14):3560-3570
[34]Bonaglia MC, Giorda R, Borgatti R, et al. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet, 2001,69(2):261-268
[35]Verhoeven WM, Egger JI, Willemsen MH, et al. Phelan-McDermid syndrome in two adult brothers: atypical bipolar disorder as its psychopathological phenotype? Neuropsychiatr Dis Treat, 2012,8:175-179
[36]Verhoeven WM, Egger JI, Cohen-Snuijf R, et al. Phelan-McDermid syndrome: clinical report of a 70-year-old woman. Am J Med Genet A, 2013,161A(1):158-161
[37]Misceo D, Rodningen OK, Baroy T, et al. A translocation between Xq21.33 and 22q13.33 causes an intragenic SHANK3 deletion in a woman with Phelan-McDermid syndrome and hypergonadotropic hypogonadism. Am J Med Genet A, 2011,155A(2):403-408
[38]Luciani JJ, de Mas P, Depetris D, et al. Telomeric 22q13 deletions resulting from rings, simple deletions, and translocations: cytogenetic, molecular, and clinical analyses of 32 new observations. J Med Genet, 2003,40(9):690-696
[39]Zollino M, Murdolo M, Marangi G, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C Semin Med Genet, 2008,148C(4):257-269
[40]Thomas NS, Durkie M, Potts G, et al. Parental and chromosomal origins of microdeletion and duplication syndromes involving 7q11.23, 15q11-q13 and 22q11. Eur J Hum Genet, 2006,14(7):831-837
[41]Schmeisser MJ, Ey E, Wegener S, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature, 2012,486(7402):256-260
[42]Shcheglovitov A, Shcheglovitova O, Yazawa M, et al.SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature, 2013,503(7475):267-271
[43]Barajas-Barajas LO, Valdez LL, Gonzalez JR, et al. Sensorineural deafness in two infants: a novel feature in the 22q distal duplication syndrome. Cardinal signs in trisomies 22 subtypes. Genet Couns, 2004,15(2):167-173
[44]Feenstra I, Koolen DA, Van der Pas J, et al. Cryptic duplication of the distal segment of 22q due to a translocation (21;22): three case reports and a review of the literature. Eur J Med Genet, 2006,49(5):384-395
[45]Jafri F, Fink J, Higgins RR, et al. 22q13.32 deletion and duplication and inversion in the same family: a rare occurrence. ISRN Pediatr, 2011,2011:829825
[46]Peeters H, Vermeesch J, Fryns JP. A cryptic duplication 22q13.31 to qter leads to a distinct phenotype with mental retardation, microcephaly and mild facial dysmorphism. Genet Couns, 2008,19(4):365-371
[47]Jamsheer A, Smyk M, Wierzba J, et al. Subtle familial translocation t(11;22)(q24.2;q13.33) resulting in Jacobsen syndrome and distal trisomy 22q13.3: further details of genotype-phenotype maps. J Appl Genet, 2008,49(4):397-405
[48]Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet, 2008,82(2):477-488
[49]Gauthier J, Champagne N, Lafreniere RG, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci U S A, 2010,107(17):7863-7868
[50]Gauthier J, Spiegelman D, Piton A, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet, 2009,150B(3):421-424
[51]Schaaf CP, Sabo A, Sakai Y, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum Mol Genet, 2011,20(17):3366-3375
[52]Waga C, Okamoto N, Ondo Y, et al. Novel variants of the SHANK3 gene in Japanese autistic patients with severe delayed speech development. Psychiatr Genet, 2011,21(4):208-211
[53]Soorya L, Kolevzon A, Zweifach J, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol Autism, 2013,4(1):18
[54]Durand CM, Perroy J, Loll F, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry, 2012,17(1):71-84
[55]Moessner R, Marshall CR, Sutcliffe JS, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet, 2007,81(6):1289-1297
[56]Ching TT, Maunakea AK, Jun P, et al. Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3. Nat Genet, 2005,37(6):645-651
[57]Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature, 2010,466(7303):253-257
[58]Wang X, McCoy PA, Rodriguiz RM, et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet, 2011,20(15):3093-3108
[59]Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev, 2012,22(3):229-237
[60]Verpelli C, Dvoretskova E, Vicidomini C, et al. Importance of Shank3 protein in regulating metabotropic glutamate receptor 5 (mGluR5) expression and signaling at synapses. J Biol Chem, 2011,286(40):34839-34850
[61]Bangash MA, Park JM, Melnikova T, et al. Enhanced polyubiquitination of Shank3 and NMDA receptor in a mouse model of autism. Cell, 2011,145(5):758-772
[62]Han K, Holder JJ, Schaaf CP, et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature, 2013,503(7474):72-77
[63]Bockers TM, Segger-Junius M, Iglauer P, et al. Differential expression and dendritic transcript localization of Shank family members: identification of a dendritic targeting element in the 3' untranslated region of Shank1 mRNA. Mol Cell Neurosci, 2004,26(1):182-190
[64]Uchino S, Wada H, Honda S, et al. Direct interaction of post-synaptic density-95/Dlg/ZO-1 domain-containing synaptic molecule Shank3 with GluR1 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor. J Neurochem, 2006,97(4):1203-1214
[65]Schaaf CP, Zoghbi HY. Solving the autism puzzle a few pieces at a time. Neuron, 2011,70(5):806-808
[66]State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci, 2011,14(12):1499-1506
[67]Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ, 2012,61(3):1-19
[68]Zou XB(邹小兵). 孤独症谱系障碍的研究进展. J Clin Pediatr(临床儿科杂志), 2010, 28(8):715-717
[69]Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res, 2011,1380:42-77
(本文编辑:张崇凡)
10.3969/j.issn.1673-5501.2014.04.014
国家自然科学基金课题:SHANK3基因缺失/点突变与孤独症表型关系的研究及机制探讨(2013NSFC:81371270)
1 复旦大学附属儿科医院 上海,201102;2 美国杜克大学医学院
徐秀,E-mail:xuxiu@shmu.edu.cn
2014-06-03
2014-07-15)
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国临床解剖学杂志(2022年3期)2022-06-06
河南大学学报(医学版)(2021年1期)2021-11-26
海外星云(2021年6期)2021-10-14
中国生殖健康(2020年4期)2021-01-18
中西医结合肝病杂志(2020年2期)2020-10-27
中国生殖健康(2018年4期)2018-11-06
医学研究杂志(2015年12期)2015-06-10
中国医科大学学报(2015年10期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
