
GRK5对大鼠星形胶质细胞活化的作用及其机制研究*
2014-08-08 07:24王莉莉马士程贺茂林
中国病理生理杂志 2014年4期
张 运, 王莉莉, 赵 茜, 马士程, 贺茂林
(首都医科大学附属北京世纪坛医院神经内科,北京 100038)
星形胶质细胞(astrocyte, AS) 是中枢神经系统具有免疫吞噬功能的细胞, 它在致炎因素作用下被激活。活化的AS既具有保护神经元的作用, 又能分泌细胞毒因子、炎症因子、补体蛋白而损害神经元。近年来, 神经胶质细胞功能受到研究者的普遍重视[1-2]。 它与多种疾病的发生发展密切相关, 如阿尔茨海默病、脑血管病、帕金森氏病等,大多数学者认为AS活化引起的炎症反应、氧化应激可能是上述疾病的重要病理机制之一[3-4]。核因子κB(nuclear factor-kappa B, NF-κB)是一种广泛存在于体内多种细胞的核转录因子, 参与多种疾病病理生理、多种基因调控、机体免疫应答和细胞凋亡的信号转导过程[5]。大量研究证实,NF-κB可通过活化星形胶质细胞与小胶质细胞,诱发炎症反应与氧化应激,从而引起神经元损伤、凋亡[5-6]。G蛋白偶联受体激酶5 (G-protein-coupled receptor kinase 5, GRK5) 是调节G蛋白偶联受体(G-protein-coupled receptors,GPCRs)的一类丝苏氨酸激酶, 其主要功能是使激活的GPCRs 脱敏, 从而中止后者介导的信号转导通路[7]。近年研究报道,GRK5能通过诱导NF-κB抑制剂IκBα的高表达而抑制NF-κB[8]。本实验通过研究对大鼠皮层星形胶质细胞GRK5基因沉默后NF-κB表达变化及其与胶质细胞活化、炎症反应和氧化应激的关系,探讨GRK5对NF-κB相关胶质细胞活化产生影响的可能机制,为神经炎性疾病研究提供实验依据。
材 料 和 方 法
1 材料
新生24 h内的Wistar大鼠(SPF级), 由北京大学医学部实验动物中心提供。DMEM/F12培养基、胎牛血清购自Gibco。N-乙酰半胱氨酸(N-acetylcysteine, NAC)、DMSO、MgATP和HEPES购自Sigma。BCA蛋白定量试剂盒购自Pierce。硝酸纤维素膜购自Millipore。GRK5抗体和IκBα抗体购自Santa Cruz。、胶质细胞原纤维酸性蛋白(glial fibrillary acidic protein, GFAP)抗体、活化半胱氨酸天冬氨酸蛋白酶3(caspase-3)抗体、 p65抗体及β-actin抗体购自Sigma。Ⅱ抗购自北京中杉金桥生物技术有限公司。 LipofectamineTMRNAiMAX购于Invitrogen。大鼠肿瘤坏死因子α (tumor necrosis factor α, TNF-α) ELISA 试剂盒购自BD Pharmigen; 一氧化氮(nitric oxide, NO)检测试剂盒购自Promega。超氧化物歧化酶(superoxide dismutase, SOD)试剂盒为南京建成生物工程研究所产品。
2 体外培养大鼠皮层星形胶质细胞
消毒断头, 取出大脑, 剥去软脑膜, 分离出大脑皮层, 剪碎后用0.125%胰酶消化,将细胞数调整为(3~5)×108/L 种植于培养瓶内, 在37 ℃、5% CO2条件下培养, 每3~4 d换液1次, 见细胞长成致密单层后进行传代培养、筛选、纯化, 用免疫组化法检测GFAP阳性细胞达95%以上时可进行实验检测[9]。
3 实验分组
3.1GRK5基因沉默 将细胞接种于涂有多聚赖氨酸盖玻片的24孔培养板,并加入DMEM培养液进行siRNA转染实验。4 μL LipofectamineTMRNAiMAX和5 μL 20 μmol/L siRNA分别加入100 μL Opti-MEM中并静置5 min。将2种溶液混合后再于室温静置20 min。将混合液加入培养的细胞中, 混匀。采用Western blotting检测siRNA干扰效率。GRK5 siRNA以及control siRNA由Dharmacon公司合成。GRK5 siRNA的干扰序列为 5’-AAG CCG UGC AAA GAA CUC UUU-3’; control siRNA的干扰序列为 5’-AAU UCU CCG AAC GUG UCA CGU-3’。
3.2NAC干预 参照文献用含5 mmol/L NF-κB抑制剂NAC的DMEM培养液培育24 h,给予置换培养液[10]。
3.3分组 按照RNA干扰沉默GRK5与加入NF-κB抑制剂NAC干预的不同,可分为4组:(1)control siRNA处理的对照组;(2)control siRNA+NAC的NF-κB抑制组;(3)GRK5 siRNA处理的GRK5 siRNA组;(4)GRK5 siRNA处理+NAC的GRK5 siRNA+NF-κB抑制组。
4 培养细胞分泌TNF-α和NO浓度的检测
收集各组细胞培养的上清,离心去除细胞碎片,采用双抗体夹心ELISA 法, 按TNF-α的ELISA 检测试剂盒说明检测上清中TNF-α浓度。采用常规Griess法测定培养细胞上清中NO浓度, 在检测前离心去除细胞碎片,按NO的检测试剂盒说明进行操作检测。标准溶液用DMEM(+10%FBS) 溶液进行稀释和配制,浓度为(μmol/L): 0、1、2、5、10、20、40、60和100,样品为细胞培养液上清。每孔分别加上述溶液、Griess reagent Ⅰ和Griess Reagent Ⅱ溶液各50 μL。即刻用酶标仪在540 nm 波长处测定吸光度值。绘制标准曲线,然后根据标准曲线得出相应的数值。
5 SOD活性的测定
作用终止后,吸去培养液, 以冰生理盐水洗涤细胞2次, 用细胞刮板刮下细胞, 冰生理盐水收集, 在冰浴中用超声细胞破碎仪破碎细胞, 显微镜下观察无完整细胞后, 应用自动生化分析仪和紫外分光光度计严格按照试剂盒说明测定细胞内SOD 活性, 结果分别以103U/(g protein)表示。
6 p65、TNF-α、IL-1β及诱导型NO合成酶(indu-cible nitric oxide synthase, iNOS) mRNA 的测定
利用Trizol 法提取细胞RNA, 用紫外分光光度计测量总RNA 的A260和A280值, 计算RNA的浓度和纯度。取1 μg总RNA, 以Oligo dT 为引物, 按Fermentas 逆转录试剂盒说明书要求先将mRNA 逆转录为cDNA。以1 μL cDNA 为模板, 在25 μL real-time PCR 体系, 于ABI PRISM 7000实时定量PCR仪上进行基因扩增(95 ℃ 30 s; 59 ℃ 60 s; 72 ℃ 30 s), 共35个循环; 72 ℃ 10 min。实时监测记录荧光强度。扩增完毕后进行熔解曲线分析。每个样品重复3遍。基因表达变化倍数采用2-ΔΔCt法进行比较,管家基因GAPDH作为内对照, ΔCt=Ct目的基因-Ct管家基因, ΔΔCt=ΔCt实验组-ΔCt对照组。
引物采用Primer Premier 5.0进行设计, IL-1β上游引物 5’-CTC CAT GAG CTT TGT ACA AGG-3’, 下游引物 5’-TGC TGA TGT ACC AGT TGG GG-3’, 扩增片段长度245 bp; TNF-α上游引物 5’-TGA ACT TCG GGG TGA TCG GTC-3’, 下游引物 5’-AGC CTT GTC CCT TGA AGA GAA C-3’,扩增片段长度295 bp; iNOS上游引物 5’-CCA AGA ACG TGT TCA CCA TG-3’, 下游引物 5’-GAT GTC CAG GAA GTA GGT GAG G-3’,扩增片段长度317 bp; p65上游引物 5’-CAA GTG CCT TAA TAG CAG GGC AAA-3’, 下游引物 5’-AGA GCT AGA AAG AGC AAG AGT CCA AT-3’, 扩增片段长度249 bp; GAPDH上游引物 5’-TGA AGC AGG CAT CTG AGG G-3’, 下游引物 5’-CGA AGG TGG AAG AGT GGG AG-3’, 扩增片段长度347 bp。
7 免疫荧光检测GFAP与活化caspase-3的表达
经多聚甲醛固定后的各组细胞, 采用免疫荧光染色。0.2% Triton X-100处理10 min, 并用10% 牛血清白蛋白室温孵育1 h 封闭无关抗原。加入Ⅰ抗4 ℃孵育24 h, PBS 充分漂洗后, 加入标记有荧光染料的Ⅱ抗中室温孵育1 h。Ⅰ抗包括兔来源抗GFAP抗体和羊来源抗活化caspase-3抗体 (Sigma);Ⅱ抗包括羊抗兔FITC 和兔抗羊TRITC(北京中杉金桥)。阴性对照以PBS代替I抗。DAPI(Sigma)用于染细胞核。倒置荧光显微镜采集图片。
荧光显微镜下观察。GFAP为绿色荧光,caspase-3为红色荧光。利用Image-Pro Plus图像分析系统测定所选区域caspase-3+细胞个数,并求得其占区域内总体细胞(DAPI+)百分比。
8 细胞蛋白的提取以及Western blotting检测蛋白表达
倒掉培养液, 用0.01 mol/L PBS 清洗2次后, 加上2 mL 0.01 mol/L PBS后放冰上孵育5 min, 将PBS倒掉甩干, 加入100 μL细胞裂解液(50 mmol/L Tris-HCl,pH 6.8,10 mmol/L EDTA,2% SDS,5 mmol/L DTT,0.5 mmol/L PMSF) 混匀后, 放冰上孵育15 min, 将裂解液刮下, 吸入EP管中, 12 000×g离心5 min(4 ℃)后提取上清液。BCA试剂盒定量蛋白。取等量样本总蛋白, 加入5× SDS loading buffer, 混匀后煮沸5 min, 10% SDS-PAGE电泳分离, 电转移至硝酸纤维素膜(300 V, 1 h), 含5%脱脂奶粉的TBST封闭,分别加入GRK5抗体(1∶200)、p65抗体(1∶500)、 IκBα抗体(1∶200)及β-actin(1∶5 000)Ⅰ抗, 4 ℃过夜。次日TBST洗膜, 辣根过氧化物酶标记的Ⅱ抗室温孵育1 h, TBST洗膜, ECL显示特异条带。β-actin抗体作为内参照。利用柯达数码1D软件对每个条带灰度进行定量分析。
9 统计学处理
数据以均数±标准差(mean±SD)表示。采用SPSS 11.0统计软件包处理。所有数据进行正态性检验。两组样本均数比较采用两个独立样本的t检验,多组间比较采用单因素方差分析。以P<0.05为差异有统计学意义。
结 果
1 细胞形态学观察
倒置显微镜下, 初接种的细胞呈球形, 4~6 h后开始贴壁, 伸出突起, 成片状聚集生长。随着培养时间的延长, 细胞体积逐渐增大, 突起伸长。传代3次以后,细胞形态趋于一致, 以纤维性星形胶质细胞为主。胞体相对较小, 但突起较长,见图1A。
2 RNA干扰沉默GRK5基因后其蛋白的表达
将细胞克隆接种于涂有多聚赖氨酸盖玻片的24孔培养板,并加入DMEM培养液进行siRNA转染实验。Western blotting检测发现经GRK5基因沉默后细胞内GRK5蛋白表达较对照组显著减少,见图1B。
3 免疫荧光法检测各组星形胶质细胞形态及凋亡率
对经过不同干预后的各组星形胶质细胞用免疫荧光法进行鉴定并检测其凋亡率。与对照组相比, NF-κB抑制组细胞GFAP荧光强度明显减弱,突起减少;GRK5 siRNA组细胞总数未见明显差异,但是细胞GFAP荧光强度增强,胞体肥大,突起增多;GRK5 siRNA+NAC组与对照组相比细胞形态及突起数量则无明显变化;GRK5 siRNA组与NF-κB抑制组及GRK5 siRNA+NAC组相比GFAP荧光强度增强,胞体肥大,突起增多,见图2A。
NF-κB抑制组中表达活化caspase-3的细胞显著少于对照组(P<0.05);GRK5 siRNA组活化caspase-3明显高于对照组(P<0.01);而GRK5 siRNA+NAC组表达活化caspase-3的细胞比率明显低于GRK5 siRNA组(P<0.01),与对照组无明显差异(P>0.05), 但是仍然显著高于NF-κB抑制组(P<0.01),见图2A、B。上述结果表明抑制NF-κB可使星形胶质细胞凋亡减少,而GRK5基因沉默可导致星形胶质细胞凋亡增加。尽管向GRK5 siRNA组细胞培养液中加入NF-κB抑制剂NAC能部分减少凋亡, 但是与NF-κB抑制组比较仍存在显著差异。
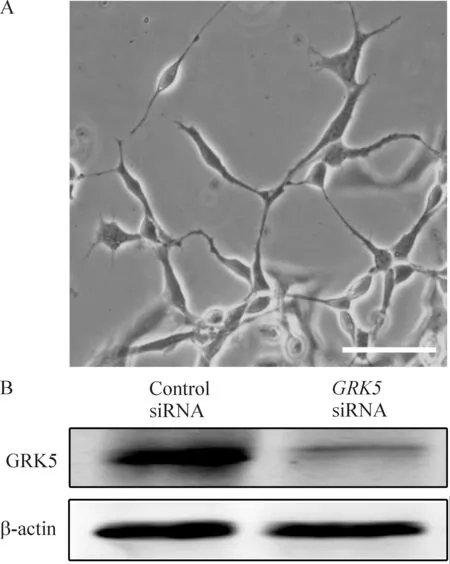
Figure 1. Morphology of the cultured astrocytes from brain cortex of newborn Wistar rats (A) and expression of GRK5 protein after GRK5 gene silencing. Scale bar=50 μm.
4 各组星形胶质细胞中p65、TBF-α、IL-1β及iNOS mRNA的表达
实时荧光定量PCR检测发现, NF-κB抑制组细胞p65 mRNA水平显著低于对照组(P<0.01);GRK5 siRNA组细胞中p65 mRNA水平显著高于对照组(P<0.01);而GRK5 siRNA+NAC组p65 mRNA水平显著低于对照组和GRK5 siRNA组(P<0.05或P<0.01),但是仍然显著高于NF-κB抑制组 (P<0.01),见图2C。这表明GRK5基因沉默可导致星形胶质细胞NF-κB表达增加。
与此同时,NF-κB抑制组细胞TNF-α、IL-1β及iNOS mRNA水平低于对照组(P<0.05或P<0.01);GRK5基因沉默可显著增加细胞中TNF-α、IL-1β及iNOS mRNA水平(P<0.01);而GRK5 siRNA+NAC组上述细胞因子mRNA水平显著低于GRK5 siRNA组(P<0.01),与对照组无明显差异(P>0.05), 但是仍然显著高于NF-κB抑制组(P<0.01),见图2C。这表明GRK5基因沉默可导致星形胶质细胞炎症因子显著升高。
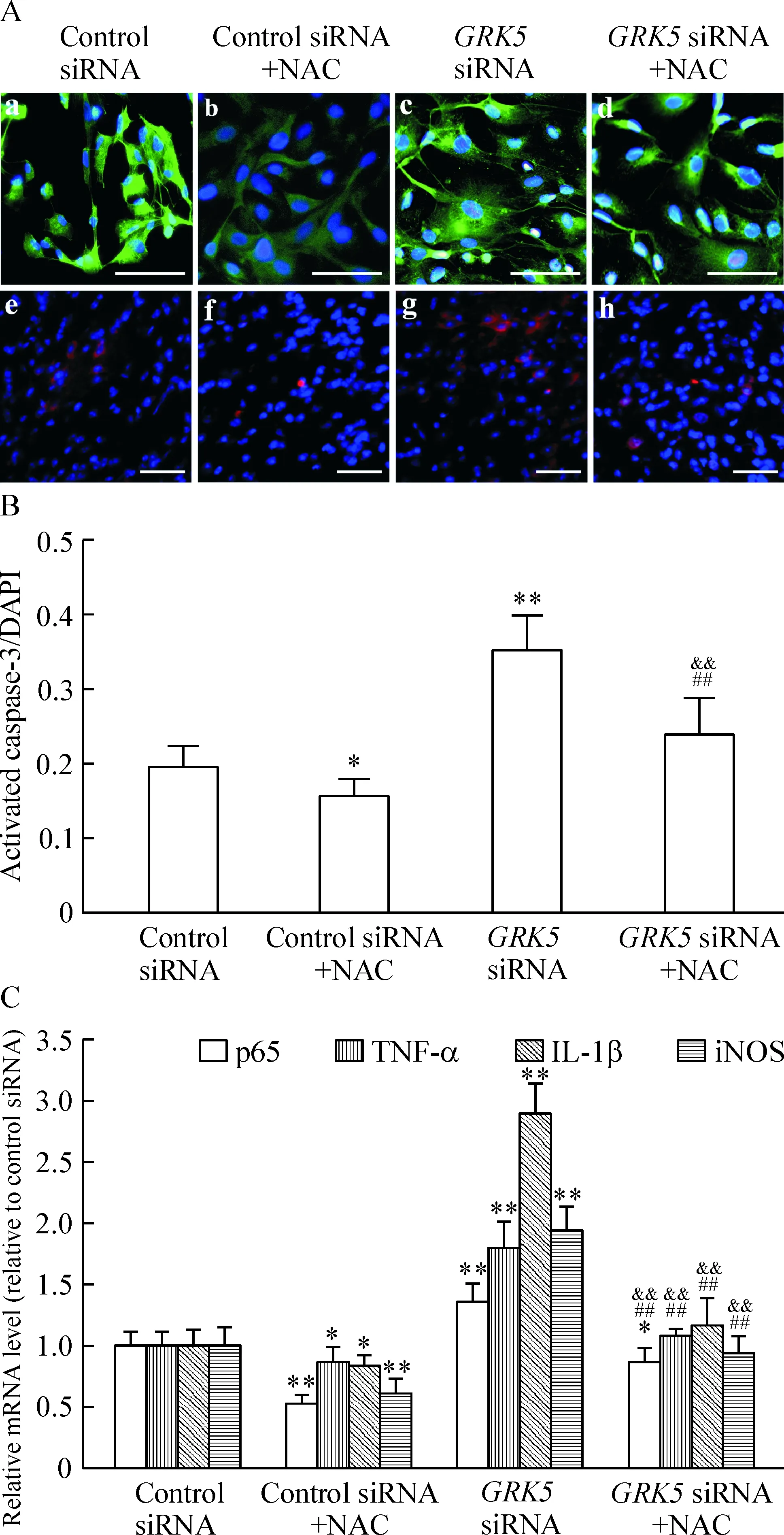
Figure 2. Identification of the cultured astrocytes, and the expression of activated caspase-3 and the mRNA levels of NF-κB p65, TNF-α, IL-1β and iNOS in the astrocytes. A: expression of GFAP (a~d) and activated caspase-3 (e~h) detected by immunofluorescence staining; B: semi-quantitative analysis of the ratio of activated caspase-3-positive cells to DAPI-positive cells; C: the mRNA levels of NF-κB p65, TNF-α, IL-1β and iNOS detected by real-time PCR. Scale bar=50 μm. Mean+SD. n=6. *P<0.05, **P<0.01 vs control siRNA group; &&P<0.01 vs GRK5 siRNA group; ##P<0.01 vs control siRNA +NAC group.
5 各组星形胶质细胞分泌TNF-α、NO浓度的检测与SOD活性的测定
ELISA检测发现,对照组的星形胶质细胞分泌处于较低水平的TNF-α和NO;NF-κB抑制剂NAC能降低TNF-α和NO分泌 (P<0.01);GRK5基因沉默后细胞分泌TNF-α和NO水平显著高于对照组(P<0.01);虽然GRK5 siRNA+NAC组TNF-α和NO水平与GRK5 siRNA组相比均显著降低(P<0.01),但仍然高于对照组(TNF-αP<0.01, NOP<0.05),且显著高于NF-κB抑制组(P<0.01),见图3A、B,可见GRK5基因沉默可以刺激星形胶质细胞分泌NO和TNF-α。

Figure 3. Effects of GRK5 siRNA on the secretion of TNF-α (A) and NO (B) and the activity of SOD (C) in astrocytes. Mean±SD. n=6. *P<0.05, **P<0.01 vs control siRNA group; &P<0.05, &&P<0.01 vs GRK5 siRNA group; ##P<0.01 vs control siRNA+NAC group.
酶活性检测发现,NF-κB抑制组SOD活性明显高于对照组(P<0.01);GRK5基因沉默后细胞SOD活性显著低于对照组(P<0.01);虽然GRK5 siRNA+NAC组SOD活性与GRK5 siRNA组相比明显升高(P<0.05),但仍显著低于对照组(P<0.01)与NF-κB抑制组(P<0.01),见图3C,可见GRK5基因沉默可抑制星形胶质细胞内SOD活性。
6 各组细胞中GRK5、p65及IκBα的表达
Western blotting检测发现NF-κB抑制剂NAC处理胶质细胞后不影响GRK5表达, 但可导致IκBα表达增加及p65表达减少(P<0.01)。GRK5基因沉默后GRK5表达低于对照组,其差异有统计学意义,同时IκBα蛋白表达较对照组显著降低, p65蛋白表达较之对照组显著增加(P<0.01)。与GRK5基因沉默组比较,GRK5 siRNA+NAC组GRK5表达无明显改变。与GRK5 siRNA组相比较,GRK5 siRNA+NAC组能增加IκBα蛋白表达, 减少p65蛋白表达水平(P<0.05),其p65蛋白表达水平与对照组无明显差异(P>0.05),但二者与NF-κB抑制组比较仍存在显著差异(P<0.01),见图4。上述结果表明GRK5基因沉默可能抑制IκBα蛋白的表达,从而增加NF-κB蛋白表达。
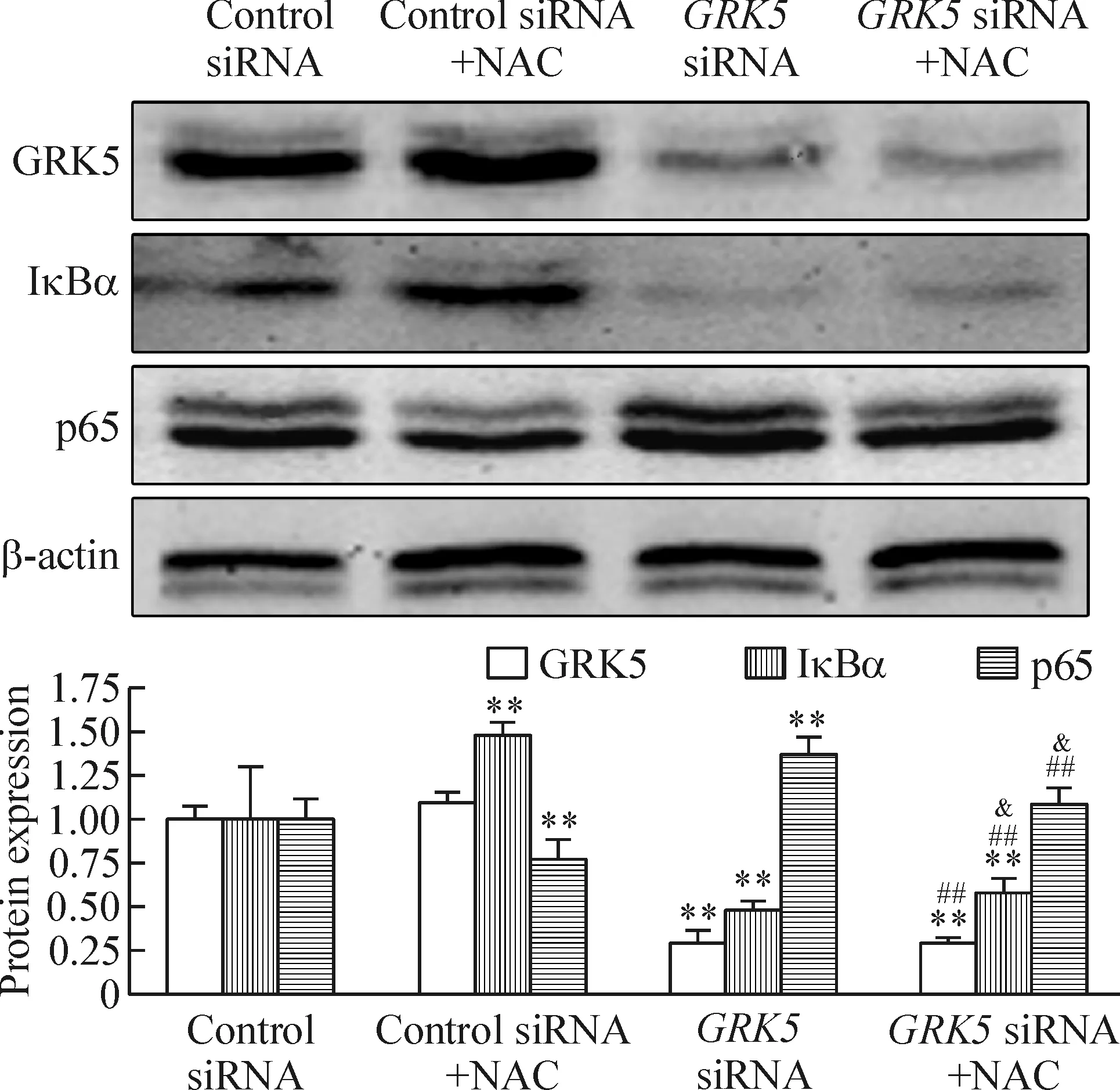
Figure 4. Western blotting assay for GRK5, IκBα and p65 protein expression. Mean±SD. n=6. **P<0.01 vs control siRNA group; &P<0.05 vs GRK5 siRNA group; ##P<0.01 vs control siRNA+NAC group.
讨 论
星形胶质细胞与神经元之间存在复杂的相互作用, 以维持神经系统内环境的稳定。当中枢神经系统遭遇炎症、缺血、损伤时,AS从静息状态快速转变为活化状态, 激活后AS对神经元的影响包括两方面: (1)通过分泌大量的神经营养因子、细胞因子和基质分子促进轴突再生, 保护神经元免受损伤, 有利于损伤后神经元的功能恢复; (2)可产生细胞炎症因子、补体、氧自由基等, 启动炎症反应、氧化应激,促进神经细胞的损伤和死亡, 加重疾病进程[11]。 而NF-κB是介导许多免疫与炎症反应的中心物质。大量研究证实,NF-κB可通过活化AS与小胶质细胞,诱发炎症反应与氧化应激,从而引起神经元损伤、凋亡[5-6]。近年研究报道,GRK5敲除小鼠老化时表现出β-淀粉样蛋白生成增加,诱发炎症反应并引起细胞凋亡[12]。更有研究发现GRK5能通过诱导IκBα表达增高,从而抑制NF-κB的表达[8],提示GRK5可能参与NF-κB激活星形胶质细胞及引起的相关反应。
已知IκBα是NF-κB的抑制蛋白。在静息状态下,染色体区域维持蛋白1(chromosome region maintenance 1, CRM-1)通过识别IκBα C末端的核转出序列(nuclear exporting sequence, NES)将IκBα/NF-κB复合体转出细胞核,NF-κB的p65和p50亚单位以失活状态存在于细胞质中[8]。当上游信号激活IκB激酶(IκB kinase,IKK)后,活化的IKK会泛素化、磷酸化并降解IκBα,使得NF-κB 2个亚单位从失活状态活化,并从细胞质转移到细胞核内(尤其是p65亚单位),与相应的炎症相关基因结合,启动炎症细胞因子转录,诱发炎症[5-6]。研究报道GRK5同源调节域能与IκBα N末端相结合,掩盖了IκBα C末端NES功能区,使得IκBα/NF-κB复合体在核内聚集,抑制了NF-κB的活化[8]。本研究通过Western blotting检测发现将培养胶质细胞GRK5基因沉默后,IκBα表达减弱,p65亚单位表达增强。该结果表明GRK5基因沉默可引起星形胶质细胞NF-κB活化,推测GRK5基因沉默解除了对IκBα C末端NES功能区的抑制,IκBα可顺利转出细胞核,易于被降解失活,从而激活NF-κB。
已有大量研究证实,TNF-α等炎症因子的分泌可活化星形胶质细胞。活化的星形胶质细胞在损伤早期也可加快释放致炎因子如TNF-α和IL-1β, 进一步激活NF-κB,加重炎症变化[5-6]。本实验荧光显微镜下观察到GRK5 siRNA组GFAP荧光强度增强、胞体肥大、突起增多,形态学上提示星形胶质细胞活化[13]。此外,实时荧光定量PCR结果显示,GRK5基因沉默刺激胶质细胞TNF-α、IL-1β和iNOS mRNA表达水平增高,结合检测到该组胶质细胞IκBα表达减弱,p65亚单位表达增多,推测与NF-κB活化刺激相关炎症因子表达增加有关[14]。收集各组细胞培养上清后,检测发现GRK5基因沉默刺激胶质细胞分泌TNF-α增多,表明GRK5基因沉默诱发了炎症反应,而炎症反应也可加剧AS活化、激活NF-κB,从而进一步分泌炎症因子。本实验还发现GRK5基因沉默刺激胶质细胞分泌NO增多,且酶活性检测结果显示,SOD活性降低。SOD是自由基清除酶,GRK5基因沉默后SOD活性下降,也反映出胶质细胞内源性抗氧化能力下降,结合检测到细胞内iNOS mRNA表达增多,表明GRK5基因沉默诱发了氧化应激反应,可能与AS活化或NF-κB活性增强相关。研究显示,活化的星形胶质细胞可诱导iNOS表达增多,产生的一氧化氮导致过亚硝酸盐生成和氧化应激反应,导致神经元损伤和凋亡[15]。亦有研究报道,NF-κB可通过上调环氧化酶2增加有害自由基的产生[16]。Caspase家族是调节细胞凋亡的关键成员, 它的级联激活引发细胞发生凋亡特异改变, 并最终崩解。Caspase-3是caspase级联反应中最关键的效应蛋白酶, 通常以酶原的形式存在, 激活后通过酶解其特异性底物, 使细胞发生凋亡[17]。本实验发现GRK5基因沉默后活性caspase-3表达增多,NF-κB抑制使得活性caspase-3表达显著减少。而GRK5基因沉默联合NAC抑制组活化caspase-3的表达明显低于GRK5 siRNA组,与对照组无明显差异, 但是仍然显著高于NF-κB抑制组。提示星形胶质细胞GRK5基因正常表达可能通过抑制caspase-3的活化而发挥抗凋亡作用。
本实验结果表明,GRK5基因沉默可能通过刺激NF-κB表达增多引起星形胶质细胞活化。推测GRK5的正常表达可通过抑制星形胶质细胞活化发挥抗炎症反应、抗氧化作用。星形胶质细胞在神经变性病的发病机制中有着不可忽视的作用,如何调控胶质细胞的功能使其向保护神经元的方向发展值得深入探索。
[参 考 文 献]
[1] Ridet JL, Malhotra SK, Privat A, et al. Reactive astrocytes: cellular and molecular cues to biological function[J]. Trends Neurosci, 1997, 20(12): 570-577.
[2] 陈 赛,蔡诗达,张 楚,等. TRPM2通道在星形胶质细胞氨中毒中的作用[J]. 中国病理生理杂志, 2013, 29(6):1039-1045.
[3] Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress[J].Biomed Pharmaother, 2004, 58(1): 39- 46.
[4] 刘付宁,纪风涛,何惠燕,等. 右美托咪定对大鼠脑缺血再灌注损伤后星形胶质细胞的影响[J].中国病理生理杂志, 2012, 28(10):1751-1755.
[5] Mattson MP, Camandola S. NF-κB in neuronal plasticity and neurodegenerative disorders[J]. J Clin Invest,2001, 107(3):247-254.
[6] Schwaninger M, Sallmann S, Petersen N, et al. Bradykinin induces interleukin-6 expression in astrocytes through activation of nuclear factor-κB[J]. J Neurochem, 1999, 73(4):1461-1466.
[7] Daigle TL, Caron MG. Elimination of GRK2 from cholinergic neurons reduces behavioral sensitivity to muscarinic receptor activation[J]. J Neurosci, 2012, 32(33):11461-11466.
[8] Sorriento D, Ciccarelli M, Santulli G, et al. The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptio-nal activity by inducing nuclear accumulation of IκBα[J]. Proc Natl Acad Sci U S A, 2008, 105(46):17818-17823.
[9] 常 利,张博爱,贾延劼,等. P2Y1受体介导Aβ25-35所致大鼠星形胶质细胞活化[J]. 中国病理生理杂志, 2011, 27(1): 67-71.
[10] Islam KN, Koch WJ. Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression[J]. J Biol Chem, 2012, 287(16):12771-12778.
[11] Kuchibhotla KV, Lattarulo CR, Hyman BT, et al. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice[J]. Science, 2009, 323(5918): 1211-1215.
[12] Suo Z, Cox AA, Bartelli N, et al. GRK5 deficiency leads to early Alzheimer-like pathology and working memory impairment[J]. Neurobiol Aging, 2007, 28(12):1873-1888.
[13] Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation[J]. Trends Neurosci, 2009,32(12):638-647.
[14] Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution[J]. FASEB J, 2006, 20(10):1589-1598.
[15] Saha RN, Pahan K. Signals for the induction of nitric oxide synthase in astrocytes[J]. Neurochem Int, 2006, 49(2):154-163.
[16] Lio D, Annoni G, Licastro F, et al. Tumor necrosis factor-alpha -308A/G polymorphism is associated with age at onset of Alzheimer’s disease[J]. Mech Ageing Dev, 2006,127(6):567-571.
[17] Yang X, Stennicke HR, Wang B, et al. Granzyme B mimics apical caspases. Description of a unified pathway for trans-activation of executioner caspase-3 and -7[J]. J Biol Chem, 1998, 273(51):34278-34283.
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
传染病信息(2022年2期)2022-07-15
昆明医科大学学报(2021年3期)2021-07-22
科学与财富(2021年33期)2021-05-10
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
初中生世界·七年级(2018年7期)2018-09-07
中国药理学与毒理学杂志(2015年3期)2015-12-16
