
硫化氢对高糖诱导的小鼠足突细胞损伤的影响*
2014-08-08 07:24卢圣霞杜月娟刘元涛傅余芹
中国病理生理杂志 2014年4期
刘 晔, 彭 力, 卢圣霞, 杜月娟, 刘元涛△, 傅余芹△
(山东大学第二医院 1肾脏内科, 2内分泌科,山东 济南 250033;3山东电力中心医院内科,山东 济南 250002; 4济南市中心医院肾脏内科,山东 济南 250013)
糖尿病肾病(diabetic nephropathy,DN)是糖尿病常见的微血管并发症之一,也是糖尿病致死致残的重要原因。近年的研究表明,足突细胞损伤在糖尿病肾病的发生尤其是蛋白尿的形成过程中具有关键作用[1]。Wnt/β-catenin是细胞内高度保守的信号通路,参与肾脏在胚胎阶段的发生发育。目前研究发现,在成人肾脏病,包括急性肾损伤、肾小球疾病、糖尿病肾病等,该途径被异常激活并参与足突细胞损伤的病理过程[2]。紧密连接蛋白2(zonula occludens-2,ZO-2)属于膜相关鸟苷酸激酶(membrane-associated guanylate kinase 3β,MAGUK)蛋白家族成员,作为骨架蛋白其广泛存在于体内上皮细胞的紧密连接处及增殖期细胞核内,参与多种途径的信号转导。已有研究发现ZO-2存在于肾小球足突细胞中,氧化应激反应可能影响其表达并可能参与调控Wnt/β-catenin通路[3]。
硫化氢(H2S)是继一氧化碳(carbonic oxide,CO)和一氧化氮(nitric oxide,NO)之后发现的新型气体信号分子。哺乳动物体内以L-半胱氨酸为底物,通过激活5’-磷酸吡哆醛依赖性酶:胱硫醚γ-裂解酶(cystathionine γ-lyase,CSE)和胱硫醚 β-合成酶(cystathionine β-synthase,CBS)生成H2S[4]。CBS和CSE在哺乳动物组织和细胞中分布广泛[5]。CBS主要存在于神经系统,而CSE主要分布于外周系统,尤其是肝、肾及血管系统[6]。最近,有研究发现H2S对糖尿病大鼠肾脏病变具有保护作用,其机制可能与抑制肾素血管紧张素系统(renin-angiotensin system,RAS)系统的激活有关[7]。然而,硫化氢对高糖导致的足突细胞损伤是否具有保护作用?目前尚未见报道。本文拟观察硫化氢在高糖诱导的肾小球足突细胞损伤中的作用,并探讨其机制。
材 料 和 方 法
1 材料
小鼠足细胞系MPC5由山东大学医学院易凡教授惠赠。无糖RPMI-1640培养基和新生胎牛血清,D-葡萄糖,CSE特异性抑制剂DL-炔丙基甘氨酸(DL-propargylglycine,PPG)及NaHS;兔抗ZO-2、兔抗CSE、小鼠抗β-catenin、抗nephrin、抗β-actin及辣根过氧化物酶标记的羊抗兔和羊抗鼠II抗,ECL发光液,BCA试剂盒,胶片。
2 主要方法
2.1细胞培养 将MPC5细胞接种于含5.5 mmol/L葡萄糖、1×104U/L γ-干扰素、10%胎牛血清、1×105U/L青霉素和100 mg/L链霉素的RPMI-1640完全培养基中,33 ℃、5% CO2培养,在γ-干扰素的作用下传代增生。继而置于不含γ-干扰素的RPMI-1640完全培养基中,37 ℃、5% CO2培养10~14 d使其分化成熟,2~3 d换液,待细胞融合至80%左右,无血清培养24 h,使细胞同步化。同步化细胞分组:高糖组(30 mmol/L葡萄糖);正常糖组:5.5 mmol/L葡萄糖+24.5 mmol/L甘露醇;正常糖(5.5 mmol/L葡萄糖)+24.4 mmol/L甘露醇+PPG(0.1 mmol/L);正常糖(5.5 mmol/L葡萄糖)+23.5 mmol/L甘露醇+PPG(1 mmol/L);正常糖(5.5 mmol/L葡萄糖)+14.5 mmol/L甘露醇+PPG(10 mmol/L);高糖(30 mmol/L葡萄糖)+ NaHS(100 μmol/L)组。于各处理时点,收集细胞并进行Western blotting分析。
2.2Western blotting 细胞经处理后,用PBS冲洗2遍,冰上裂解,裂解液:20 mmol/L Tris-HCl, 0.1 mmol/L Na3VO4,25 mmol/L NaF,25 mmol/L β-磷酸甘油,2 mmol/L EDTA,2 mmol/L EGTA,1 mmol/L DTT,1 mmol/L PMSF,2 mg/L 抑肽酶(aprotinin),2 mg/L亮抑蛋白酶肽(leupeptin),pH 7.5。4 ℃条件下离心15 min(14 000 r/min),取上清并用BCA法测定蛋白浓度。取等量蛋白(20 μg)上样,经10%SDS聚丙烯酰胺凝胶电泳后转至PVDF膜。5%脱脂牛奶封闭1 h,4 ℃条件下,I抗孵育12 h,TBST充分洗膜后,1∶2 000稀释的辣根过氧化物酶标记II抗室温杂交1 h。TBST洗膜,ECL显色,结果定量分析用NIH ImageJ 1.42软件处理。
3 统计学处理
采用SPSS 17.0统计分析。数据以均数±标准差(mean±SD)表示,组间比较采用单因素方差分析(One-way ANOVA),两两比较用Newman-Keuls检验。以P<0.05为差异有统计学意义。
结 果
1 高糖对足突细胞nephrin、ZO-2和β-catenin表达的影响
高糖(30 mmol/L)处理12 h,足突细胞nephrin及ZO-2蛋白水平显著降低,48 h降至最低;β-catenin蛋白水平12 h显著升高(P<0.05),48 h达到高峰(P<0.01),见图1。
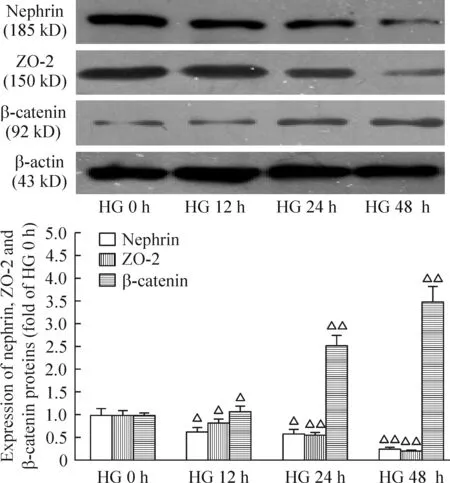
Figure 1. Effects of high glucose (HG,30 mmol/L) on nephrin,ZO-2 and β-catenin protein expression at different time points. Mean±SD. n=3. △P<0.05,△△P<0.01 vs HG 0 h group.
2 高糖对CSE蛋白表达的影响
与NG组相比,HG组CSE的表达量明显下降(P<0.05),48 h降至最低(P<0.01),见图2。
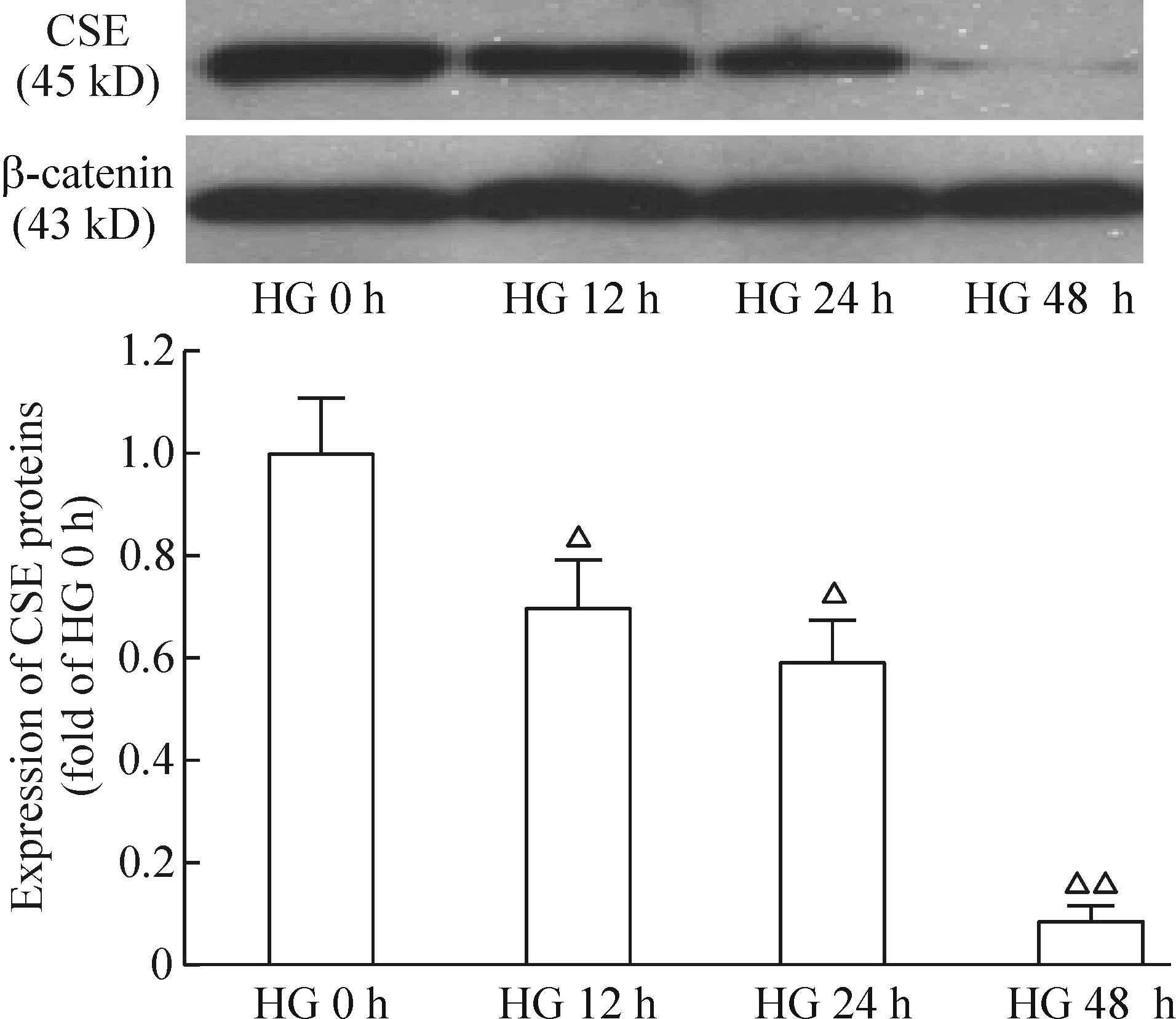
Figure 2. The effect of high glucose (HG,30 mmol/L) on expression of CSE at 0 h,12 h,24 h and 48 h. Mean±SD. n=3. △P<0.05,△△P<0.01 vs HG 0 h group.
3 PPG对小鼠足突细胞nephrin、ZO-2及β-catenin的影响
对正常糖浓度培养的足突细胞,分别给予不同浓度的PPG处理48 h,PPG可显著抑制nephrin和ZO-2的表达(P<0.05),并具有浓度依赖性;而同时显著增加β-catenin的蛋白表达水平(P<0.05),见图3。
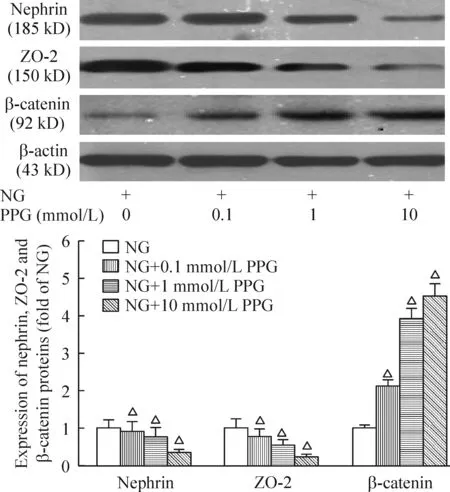
Figure 3. Effects of different concentrations of PPG on the expression of nephrin,ZO-2 and β-catenin at 48 h. Mean±SD. n=3. △P<0.05 vs NG group.
4 NaHS对高糖培养的足突细胞nephrin、ZO-2及β-catenin表达的影响
与NG组相比,HG处理48 h nephrin和ZO-2的表达量明显减少(P<0.01),β-catenin的表达量明显增加(P<0.01);HG+NaHS组nephrin和ZO-2蛋白水平较HG组显著增加(P<0.01),但仍低于NG组(P<0.01);β-catenin较HG明显减少(P<0.01),但仍高于NG组(P<0.01),见图4。
讨 论
蛋白尿是糖尿病肾病重要的临床表现,是临床诊断的重要指标,从早期的微量白蛋白尿到中晚期的大量蛋白尿均可使肾功能进行性恶化。糖代谢异常是糖尿病肾病足突细胞病变的始动因素,高血糖可以使肾小球滤过膜的通透性增加[8],足突细胞是肾小球滤过膜的最后一道屏障,兼有大小选择性和电荷选择性,其病变可导致大分子血浆蛋白从肾小球漏出,大量蛋白的漏出又会加重足突细胞损伤和肾小球硬化。足突细胞的脱落和凋亡明显落后于蛋白尿的产生,这提示受高糖刺激后,足突细胞分子水平损伤的触发最后导致蛋白尿的产生。Nephrin由NPHS1编码,是足突细胞裂孔膜的标记蛋白之一,与白蛋白从滤过膜的滤出密切相关,其下调还可进一步导致肾小球硬化和肾功能恶化[9],是足突细胞损伤的重要标志性蛋白,已有研究表明其下调可能与Wnt/β-catenin通路的异常激活有关[10]。
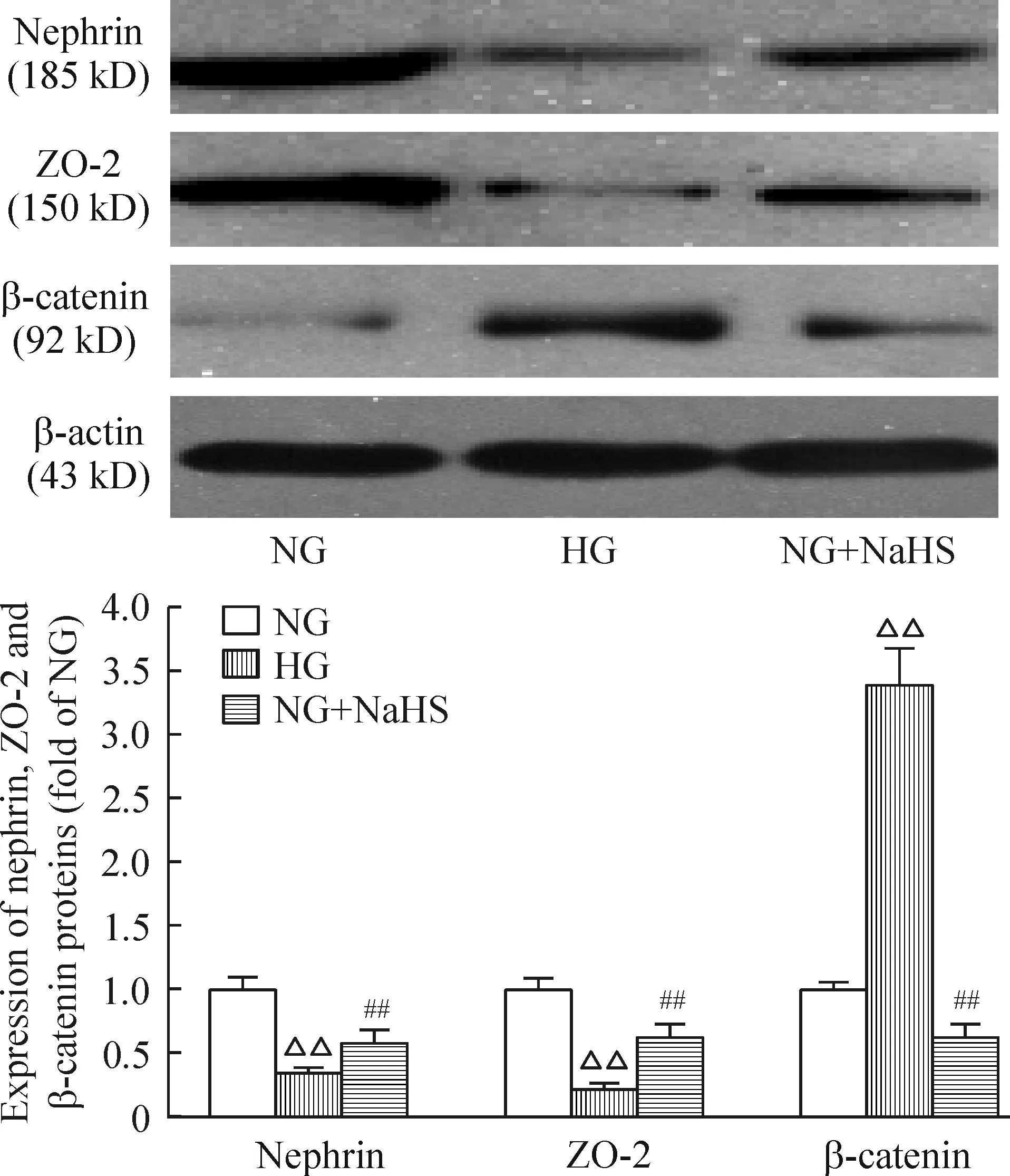
Figure 4. The effect of hydrogen sulfide on protein levels of nephrin, ZO-2 and β-catenin at 48 h. Mean±SD. n=3. △△P<0.01 vs NG group;##P<0.01 vs HG group.
Wnt信号通路在足突细胞损伤和蛋白尿形成中起关键作用,在Wnt缺乏时,转录激活因子和黏着连接的连接蛋白β-catenin与轴蛋白、大肠腺瘤样息肉蛋白和糖原合成酶激酶3β(glycogen synthase kinase 3β,GSK3β)结合成复合体存在于胞质中,GSK3β可以磷酸化β-catenin并使其降解,细胞内β-catenin含量较低;当该通路被激活时,作为胞外配体的Wnt与细胞表面的Frizzled受体和共受体低密度脂蛋白受体相关蛋白结合,GSK3β被磷酸化失活,β-catenin无法被泛素化而不能降解,胞浆中富集的β-catenin移位至核内,与转录因子LEF/TCF一起激活靶基因的转录。在足突细胞中,如Wnt1等多种Wnt家族蛋白表达的增多,抑制了β-catenin的磷酸化,胞内富集的β-catenin可活化表达关键的转录因子snail,进而抑制nephrin的表达导致足突细胞功能障碍[11-12],最新研究显示,ZO-2可能参与调控Wnt通路[3,13]。
ZO-2是广泛表达在上皮细胞紧密连接处及核内的骨架蛋白,带有多个膜相关鸟苷酸激酶特征性的蛋白结合位点,包括3个PDZ结构域,1个SH3结构域,1个GuK结构域[3],作用广泛,比如,可以将紧密连接闭合蛋白和封闭蛋白结合在肌动蛋白骨架上,能与黏着连接的α-连环蛋白、缝隙连接蛋白36和43以及其它紧密连接蛋白如ZO-1、扣带蛋白结合。在肾脏,ZO-2存在于肾小球足突细胞和肾小管上皮细胞中,与ZO-1不同的是,在肾小球ZO-2与巢蛋白(nestin)共定位于足突细胞的足突上,目前已知其与体内多种细胞增殖、凋亡、肿瘤和氧化应激相关[13-14]。有研究指出,ZO-2可抑制GSK3β被磷酸化,促进β-catenin在胞质中的累积,防止nephrin的丢失,避免足突细胞大规模融合或丢失[3,13]。这可能是足突细胞功能障碍导致蛋白尿形成的重要机制之一。
肾脏的内源性H2S通过半胱氨酸由CBS、CSE和3-巯基丙酮酸转硫酶催化作用产生[15],具有强大的抗氧化、抗凋亡和抗炎作用,Yuan[16]等研究发现,硫化氢可以改善高糖条件下产生的活性氧簇(reactive oxygen species,ROS)对肾小球系膜细胞的损伤,高糖环境影响降低NAPDH氧化酶这类促进ROS产生的酶类活性,打破了氧化还原反应的平衡,使硫化氢生成减少,足突细胞是否含有合成内源性H2S的酶类及其对足细胞的作用机制目前仍不清楚。
本研究发现高糖培养的足突细胞表达nephrin明显减少,提示高糖可导致足突细胞的损伤,我们继而观察了ZO-2和β-catenin的变化,结果表明高糖刺激显著降低了ZO-2的表达,激活了Wnt/β-catenin通路,这与既往研究结果一致。CSE是体内产生硫化氢所必需的酶类,业已证实其在肾脏中高表达,细胞水平上已证实系膜细胞中存在CSE,而足突细胞中是否存在还未证实。我们研究证实在足突细胞中有CSE表达,此外,本实验结果显示高糖在抑制nephrin表达的同时,也降低了CSE的表达水平。为了明确高糖诱导的CSE降调节是否与足突细胞损伤有关, 我们进一步观察了CSE特异性抑制剂PPG对nephrin表达的影响。结果发现CSE特异性抑制剂显著降低了正常糖条件下的小鼠足突细胞的nephrin表达,且具有剂量依赖性,从而提示高糖诱导的CSE降调节可能是造成足突细胞损伤的机制之一。此外,我们以100 μmol/L NaHS[17-18]作为硫化氢的供体处理细胞,结果显示NaHS对高糖诱导的Nephrin降调节具有显著抑制作用,提示外源性硫化氢对高糖环境下的小鼠足突细胞损伤具有一定的防治作用。
为了探讨硫化氢的作用机制,我们观察了CSE特异性抑制剂对ZO-2及Wnt/β-catenin通路的影响,结果提示CSE特异性抑制剂可显著抑制ZO-2表达,而增加β-catenin的蛋白水平。与之相反,外源性硫化氢则显著抑制了高糖诱导的ZO-2降调节及β-catenin蛋白水平的升高。以上结果提示,硫化氢对高糖诱导足突细胞损伤的保护作用可能是通过对ZO-2蛋白的表达调控而实现的,但详细机制尚有待进一步探讨。总之,本研究结果提示高糖诱导的足突细胞CSE表达降低可能是足突细胞损伤的重要机制之一。而外源性硫化氢对高糖诱导的足突细胞损伤具有一定保护作用,其机制可能与增加ZO-2蛋白表达、抑制Wnt/β-catenin通路有关。本研究为糖尿病肾病的防治提供了新的思路,而本研究结果有待体内实验进一步证实。
[参 考 文 献]
[1] 覃乔静,邓华聪,曹文富. 糖尿病肾病肾小球nephrin表达的变化[J]. 中国病理生理杂志,2007,23(2):391-392.
[2] 周 静,袁伟杰,谢院生,等. siRNA沉默WT1对小鼠足细胞Wnt/β-catenin和nephrin表达的影响[J].中国病理生理杂志,2013,29(2):219-224.
[3] Gonzalez-Mariscal L,Bautista P,Lechuga S,et al. ZO-2,a tight junction scaffold protein involved in the regulation of cell proliferation and apoptosis[J]. Ann N Y Acad Sci,2012,1257:133-141.
[4] Moore PK Bhatia M,MoochhaIa S. Hydrogen sulfide:from the smell of the past to the mediator of the future?[J]. Trends Pharmacol Sci,2003,24(12):609-611.
[5] Kamoun P. Endogenous production of hydrogen sulfide in mammals[J]. Amino Acids,2004,26(3):243-254.
[6] Ishii I,Akahoshi N,Yu XN,et al. Murine cystathionine γ-lyase:complete cDNA and genomic sequences,promoter activity,tissue distribution and developmental expression[J]. Bioehem J,2004,381(Pt 1): 113-123.
[7] Xue H,Yuan P,Ni J,et al. H2S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation[J]. PLoS One,2013,8(9):e74366.
[8] Sward P,Rippe B. Acute and sustained actions of hyperglycaemia on endothelial and glomerular barrier permeability[J]. Acta Physiol (Oxf),2012,204(3):294-307.
[9] 梁 伟,任志龙,韦忠平,等. Nephrin稳定血管紧张素II诱导的足细胞细胞骨架改变的机制研究[J].中国病理生理杂志,2012,28(12):2216-2221.
[10] Dai C,Stolz DB,Kiss LP,et al. Wnt/β-catenin signaling promotes podocyte dysfunction and albuminuria[J]. J Am Soc Nephrol,2009,20(9):1997-2008.
[11] He WC,Dai CS,Li YJ,et al. Wnt/β-catenin signaling promotes renal interstitial fibrosis[J]. J Am Soc Nephrol,2009,20(4):765-776.
[12] Yook JI,Li XY,Ota I,et al. Wnt-dependent regulation of the E-cadherin repressor Snail[J]. J Biol Chem,2005,280(12):11740-11748.
[13] Bautista-García P,Reyes JL,Martín D,et al. Zona occludens-2 protects against podocyte dysfunction induced by ADR in mice[J]. Am J Physiol Renal Physiol,2013,304(1):F77-F87.
[16] Yuan P,Xue H,Zhou L,et al. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide[J]. Nephrol Dial Transplant,2011,26(7):2119-2126.
[17] Abe K,Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator[J]. J Neurosci,1996,16(3):1066-1071.
[18] Wang R. Two’s company,three’s a crowd: can H2S be the third endogenous gaseous transmitter?[J]. FASEB J,2002,16(13):1792-1798.
猜你喜欢
昆明医科大学学报(2021年12期)2021-12-30
石油沥青(2021年5期)2021-12-02
现代临床医学(2021年5期)2021-11-02
天津医科大学学报(2021年4期)2021-08-21
能源工程(2021年1期)2021-04-13
老友(2021年3期)2021-03-28
家庭百事通·健康一点通(2020年11期)2020-11-30
中国盐业(2018年12期)2018-09-21
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
