
新发基因突变致成人线粒体神经胃肠脑肌病
2014-07-19 12:01冯云路朱以诚钱家鸣
协和医学杂志 2014年3期
谭 蓓,冯云路,吴 东,朱以诚,陈 钰,钱家鸣
中国医学科学院 北京协和医学院 北京协和医院 1消化内科 2神经科 3放射科, 北京100730
·论 著·
新发基因突变致成人线粒体神经胃肠脑肌病
谭 蓓1,冯云路1,吴 东1,朱以诚2,陈 钰3,钱家鸣1
中国医学科学院 北京协和医学院 北京协和医院1消化内科2神经科3放射科, 北京100730
目的 探讨线粒体神经胃肠脑肌病患者的临床表现及基因突变情况。方法 分析1例成人线粒体神经胃肠脑肌病患者临床资料,对患者及其家系线粒体病相关基因应用NimbleGen固相芯片进行目标区域捕获测序。结果 该患者表现为进行性加重的假性胃肠梗阻、脑白质病、恶液质、周围神经病、眼外肌无力及多种代谢紊乱。基因检测发现TYMP基因c.217G>A纯合突变为该患者的致病突变,患者父母(近亲婚配)及姐姐均为该突变杂合子,该突变为新发突变。结论 经基因检测确诊TYMP基因新发突变致成人线粒体神经胃肠脑肌病。
线粒体神经胃肠脑肌病;胸腺嘧啶核苷酸磷酸化酶基因;基因突变
MedJPUMCH,2014,5(3):302-306
线粒体负责细胞的能量合成与供应,其结构功能异常可以导致多系统损害。线粒体病是由细胞核DNA(nDNA)或线粒体DNA(mtDNA)缺陷所致的一类多系统疾病,其中以假性胃肠梗阻及外周神经病变为主要表现的一型称为线粒体神经胃肠脑肌病(mitochondrial neurogastrointestinal encephalomyopathy,MNGIE)。MNGIE是一种罕见的常染色体隐性遗传病,1994年由Hirano等[1]首次提出并命名,主要表现为胃肠道动力障碍、恶液质、渐进性眼睑下垂、眼外肌麻痹、周围神经病和脑白质病。位于染色体22q13.33的胸腺嘧啶核苷酸磷酸化酶(thymidine phosphorylase, TYMP)基因为致病基因。研究MNGIE的临床表现及遗传背景,有助于深入了解人体能量代谢的分子机制,并为此类疾病的治疗开辟新的路径。国外目前约有90例散发性或家族性MNGIE病例以及50种TYMP基因突变报道[2],国内仅有2例报道[3- 4],其中仅1例患者进行基因检测。本研究对1例成人MNGIE患者的临床表现及其家系基因情况进行了相关分析。
资料和方法
临床资料
患者男性,32岁,入院前1年余出现劳累后腹胀,剧烈呕吐大量胃液,伴腹泻,排稀糊样便5~6次/d,1周后呕吐量增至2500~3000 ml/d。不发热,无腹痛、里急后重。予禁食水、胃肠减压后症状改善,可进半流食,体重增加4 kg,情况一度稳定。6个月前再次出现恶心及大量呕吐,予静脉营养并置入空肠导管予肠内营养。2个月前患者病情加重,不能进食,体重下降15 kg,伴四肢疼痛、麻木,1个月前再次入院。既往史、个人史及婚育史无特殊。家族史:父母系近亲结婚(表兄妹)。查体:心率100~120次/min,血压95/50 mm Hg。身高167 cm,体重37 kg,体重指数13.3 kg/m2。恶液质。神志清楚,高级智能正常。双眼上睑缘遮盖瞳孔缘2~3 cm,双眼各项运动均不到位,双眼瞳孔直径5 mm,对光反射迟钝。心肺(-)。舟状腹,腹软,无压痛、反跳痛及肌紧张,肝脾肋下未及,胃振水音(+),肠鸣音2~3次/min。双上肢肘关节以下、双下肢小腿针刺觉减低,四肢肌力V级,肌张力正常,腱反射低,双侧病理征阴性。入院后查血常规白细胞27.63×109/L,中性粒细胞83%,血红蛋白87 g/L,血小板135×109/L。粪便常规及隐血(-)。生化:谷丙转氨酶67 U/L,白蛋白30 g/L,肌酐374 mol/L,尿素13.00 mmol/L,甘油三酯18.08 mmol/L,胆固醇4.93 mmol/L,肌酸激酶281 U/L。血气:酸碱度7.52,氧分压66 mm Hg,二氧化碳分压68 mm Hg,碳酸氢根55.5 mmol/L,乳酸 3.1 mmol/L。抗核抗体9项、补体、免疫球蛋白、血清蛋白电泳、免疫电泳(-),甲状腺功能(-)。血、尿重金属毒物检测(-),尿卟啉及卟胆原(-)。代谢脑病6项:芳基硫酸酯酶A、半乳糖脑苷脂酶、β半乳糖苷酶、氨基己糖苷酶A、氨基己糖苷酶(A+B)、α半乳糖苷酶均正常。空腹血糖、胰岛素、C肽(-)。上消化道造影示胃腔扩大,低张力胃,胃内较多液体潴留(图1)。腹盆CT:胃腔扩大,重度脂肪肝,脾大,全组小肠明显扩张伴肠壁增厚,腹盆腔积液(图2)。胃镜示胃腔宽大,胃壁蠕动弱。小肠镜示胃腔及十二指肠腔扩张,蠕动差,第2~4组小肠通畅。为除外中枢性呕吐行头颅磁共振成像,示脑白质对称性分布大片异常信号,部分深部核团受累,符合“脑白质病”改变(图3)。肌电图示四肢周围神经源性损害。眼底示左眼视网膜中央静脉阻塞,右眼视网膜出血。入院后予以积极肠外、肠内营养支持,补充多种维生素、叶酸、各种微量元素,加用辅酶Q10、左卡尼汀调节代谢,急性肾损伤和水电解质紊乱得以纠正,但仍四肢麻木、疼痛明显,终因经济原因放弃治疗出院,随访得知患者返家5 d后死亡。
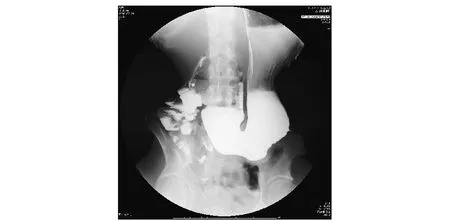
图 1 上消化道造影见胃腔扩大,蠕动减少
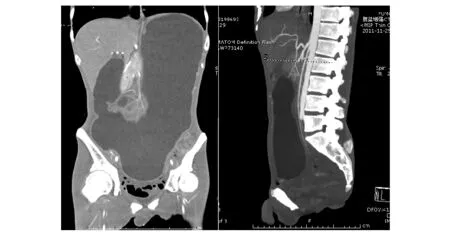
图 2 CT示胃腔扩张,肠系膜上动脉与腹主动脉夹角变小,脂肪肝
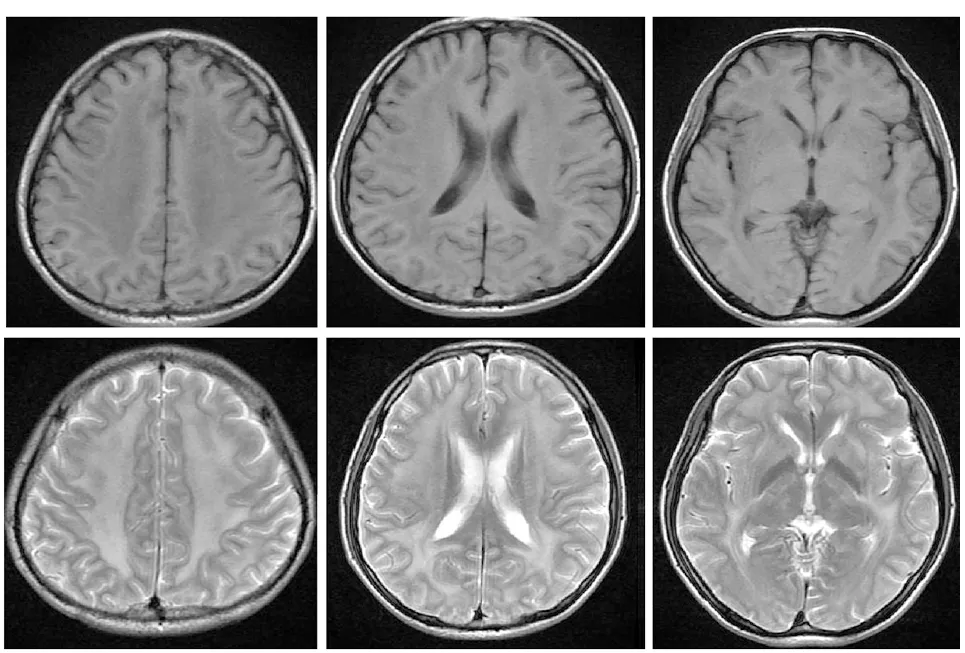
图 3 头颅磁共振成像示弥漫性脑白质病变,范围广泛,累及深部核团,但胼胝体未受累
家系情况
患者父母系表兄妹近亲结婚,家系遗传图谱见图4。
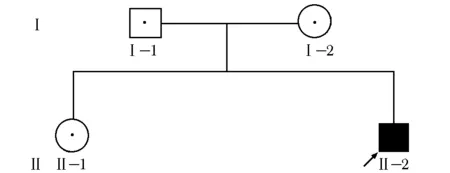
图 4 患者家系遗传图谱Ⅰ- 1.父亲;Ⅰ- 2.母亲;Ⅱ- 1.姐姐;Ⅱ- 2.患者
基因检测
对患者及其家系的线粒体基因和1056种线粒体核基因进行目标区域捕获测序。应用QIAamp DNA Blood Mini试剂盒(德国Qiagen公司)提取外周血DNA,定制线粒体基因组区域探针,与基因组DNA进行NimbleGen固相芯片(罗氏公司)杂交,将线粒体基因区域DNA富集后进行HiSeq 2000 (IIIumina公司)高通量测序,运用SOAPaligner软件对比参考基因组进行数据分析(天津华大基因公司)。
结 果
临床诊断
患者临床表现包括:(1)假性胃肠梗阻、恶液质;(2)明确的中枢脑白质病变;(3)四肢反射减低,远端感觉异常,肌电图提示周围神经损害;(4)眼睑下垂、眼外肌麻痹;结合患者父母近亲结婚史,临床诊断为MNGIE。
基因检测结果
患者存在TYMP基因的c.217G>A纯合突变,导致第73位氨基酸由丙氨酸变为苏氨酸(p.Ala73Thr),患者父亲、母亲及姐姐均为该突变杂合子,考虑为该家系的致病突变。患者MT-RNR2线粒体基因2805位轻链发生A→T杂合突变m.2805A>T(表1),而其母亲和姐姐基因中均未检测到该位点发生变异。
讨 论
线粒体病是由mtDNA或nDNA基因缺陷所致,通过母系遗传或孟德尔模式遗传。MNGIE是一种渐进性退行性多系统受累疾病,可发生于10~50岁人群,其中60%~73%患者20岁前发病[5]。核基因TYMP突变为明确致病基因。胸腺嘧啶核苷酸磷酸化酶(thymidine phosphorylase,TP)能够催化脱氧胸腺嘧啶核苷(dThd)及脱氧尿嘧啶核苷(dUrd)转变为胸腺嘧啶(TMP)及尿嘧啶(UTP),即dThd磷酸化顺向反应;dThd还能通过胸腺嘧啶激酶储存为一磷酸脱氧胸腺嘧啶核苷酸,并进一步磷酸化为二磷酸及三磷酸脱氧胸腺嘧啶核苷酸,即胸腺嘧啶脱氧核苷补救途径。mtDNA复制过程中主要依靠胸腺嘧啶脱氧核苷补救途径产生的三磷酸脱氧胸腺嘧啶核苷酸[6]。TYMP基因突变导致TP活性基本消失,催化底物dThd及dUrd蓄积,细胞质脱氧核糖核苷三磷酸(dNTP)池不平衡,线粒体dNTP池部分依赖从胞浆输入,导致线粒体dNTP失衡,损害mtDNA合成和导致不稳定。
TP广泛分布在消化道、周围神经、脑、脾、膀胱及肺,不在肌肉、肾脏、胆囊、主动脉及脂肪表达[7]。TP在胃肠道的减少导致肠壁的自主神经和平滑肌受损,45%~67%患者以胃肠道症状为主诉就诊,最常见为小肠动力下降及胃排空延迟,导致明显营养吸收障碍,并最终出现胃肠道假性梗阻和恶液质[8]。TP在周围神经的减少导致外周运动神经元脱髓鞘和感觉神经病变,可能会伴随轴突神经病,表现为双下肢对称性远端肌力下降,双手袜套样感觉异常以及刺痛、麻木、疼痛等,颅神经受累导致上睑下垂和眼肌麻痹。TP在脑的减少导致皮质神经元营养作用下降和胶质细胞增生抑制作用减轻,在此基础上线粒体损害及血脑屏障渗透性改变,可以导致白质脱髓鞘和胶质增生,引发脑白质病。生化检查可发现高乳酸MNGIE的治疗包括避免过量运动、防治感染;避免服用干扰呼吸链功能的药物,包括丙戊酸及其衍生物;积极行全肠外营养支持;补充辅酶Q10[9- 11]、维生素E以及左旋肉毒碱[11]均有利于改善代谢。此外,有研究表明血液透析或腹膜透析可清除循环中dThd及减少肾脏重吸收;反复输注富含TP的血小板可以提高TP活性,清除血浆中dThd和dUrd[2]。近年,有报道异基因造血干细胞移植(hemopoietic stem cell transplantation,HSCT)治疗后,可以观察到TP活性恢复,血dThd、dUrd浓度及乳酸下降,临床随访观察到胃肠道蠕动有所改善,但神经系统恢复不明显[12],HSCT是目前最有前景的治疗方法[13]。但总体而言,MNGIE长期预后较差,一项纳入35例患者的研究中,患者死亡年龄在26~58岁,中位生存期仅为38岁[14]。

表 1 患者致病基因检测情况
血症、脑脊液蛋白增加。临床诊断除上述症状外,主要依靠TP活性、血dThd、dUrd浓度和分子遗传学TYMP基因检测。肌肉活检有助于发现线粒体形态异常,但很遗憾本例患者拒绝行肌肉活检。
本例患者青年男性,30岁发病,以腹胀、腹泻伴呕吐起病,胃肠道动力障碍明显并进行性加重,随后出现四肢麻木疼痛,肌电图提示外周神经病变。头颅磁共振提示双侧对称性弥漫而严重的脑白质病变,无法以单纯营养不良解释。结合患者存在眼外肌麻痹、眼睑下垂,血肌酸激酶升高,与组织缺血缺氧无关的高乳酸血症[15],且存在父母近亲婚配史,考虑为MNGIE。治疗方面,积极补充多种维生素、叶酸、各种微量元素,加用辅酶Q10、左卡尼汀调节代谢。但患者仍四肢麻木疼痛明显,周围神经病变相关症状无明显改善。另一方面,患者营养问题较为突出,存在严重假性胃肠梗阻的胃肠道动力问题,治疗初期先予以肠外营养,但考虑存在重度脂肪肝、高甘油三酯血症的脂代谢异常,需应用低脂肪乳配方且适当增加糖含量以保证能量供给,同时注意避免过多的糖摄入而增加感染及渗透性腹泻的风险。当患者情况有所改善后,即置入空肠营养管努力尝试增加肠内营养。但患者胃肠道动力和外周神经病变进行性恶化,终因经济等客观原因放弃治疗自动出院,于返家后5 d死亡。
该患者核基因中存在TYMP基因的c.217G>A纯合突变,导致第73位丙氨酸替代为苏氨酸(p.Ala73Thr),患者父亲、母亲及姐姐均为该突变杂合子携带者,考虑为该家系的致病突变,且查阅国内外文献尚无该位点突变报道,为新发突变。患者线粒体基因中存在已知致病基因MT-RNR2的新突变m.2805A>T,而其母亲和姐姐基因中均未检测到该位点发生变异。TYMP基因突变会使其编码的TP缺乏,从而dThd和dUrd在循环系统中的水平升高,进一步导致mtDNA的缺失、缺陷和点突变,由此推测TYMP基因c.217G>A的纯合突变或许引发了线粒体基因MT-RNR2的m.2805A>T突变。
[1]Hirano M, Silvestri G, Blake DM, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder[J]. Neurology, 1994,44:721- 727.
[2]Libernini L, Lupis C, Mastrangelo M, et al. Mitochondrial Neurogastrointestinal Encephalomyopathy: novel pathogenic mutations in thymidine phosphorylase gene in two Italian brothers[J]. Neuropediatrics, 2012,43:201- 218.
[3]许二赫,张弥兰,董会卿,等. 线粒体神经胃肠型脑肌病的临床和病理分析[J]. 中风与神经疾病杂志, 2013,30:396- 399.
[4]唐吉刚, 李传芬, 李靖,等. 线粒体神经胃肠脑肌病一例临床、病理及基因分析[J].中华神经科杂志,2014,47:26- 29.
[5]Shoffner JM. Mitochondrial neurogastrointestinal encephalopathy disease[M]//Pagon RA. GeneReviews.Seattle:2005,22.
[6]Hirano M, Nishigaki Y, Marti R. Mitochondrial neurogastrointestinal encephalomyopathy(MNGIE): a disease of two genomes[J]. Neurologist, 2004,10:8- 17.
[7]Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder[J]. Science, 1999,283:689- 692.
[8]Teitelbaum JE, Berde CB, Nurko S, et al. Diagnosis and management of MNGIE syndrome in children: case report and review of the literature[J]. J Pediatr Gastroenterol Nutr,2002,35:377- 383.
[9]Glover EI, Martin J, Maher A, et al. A randomized trial of coenzyme Q10 in mitochondrial disorders[J]. Muscle Nerve, 2010, 42:739- 748.
[10]Chen RS, Huang CC, Chu NS. Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Short-term double-blind, crossover study[J]. Eur Neurol, 1997, 37:212- 218.
[11]DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases[J]. Muscle Nerve, 2006, 34:265- 283.
[12]Hussein E. Non-myeloablative bone marrow transplant and platelet infusion can transiently improve the clinical outcome of mitochondrial neurogastrointestinal encephalopathy: a case report[J]. Transfus Apher Sci, 2013,49:208- 211.
[13]Halter J, Schüpbach WM, Casali C,et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach[J]. Bone Marrow Transplant, 2011,46:330- 337.
[14]Nishino I, Spinazzola A, Papadimitriou A, et al. Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations[J]. Ann Neurol, 2000, 47:792- 800.
[15]Tarnopolsky M, Stevens L, MacDonald JR, et al. Diagnostic utility of a modified forearm ischemic exercise test and technical issues relevant to exercise testing[J]. Muscle Nerve, 2003, 27:359- 366.
Mitochondrial Neurogastrointestinal Encephalomyopathy Caused by A Novel TYMP Gene Mutation in An Adult Patient
TAN Bei1, FENG Yun-lu1, WU Dong1, ZHU Yi-cheng2, CHEN Yu3, QIAN Jia-ming1
1Department of Gastroenterology,2Department of Neurology,3Department of Radiology, Peking Union Medical College Hospital,Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China
WU Dong Tel: 010-69155014,E-mail:wudong061002@aliyun.com
Objective To analyze the clinical features and genetic background of mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Methods The clinical data of an adult patient with MNGIE were retrospectively reviewed. Meanwhile, the mitochondrial disease-related gene of the patient and his families were detected by target area capture sequencing with NimbleGen solid phase chip. Results This patient presented with progressive pseudo-gastrointestinal obstruction, leukoencephalopathy, cachexia, peripheral neuropathy, extraocular muscle weakness, and multiple metabolic disorders. A homozygous mutation (TYMP gene c.217G>A) was identified. The patient’s parents and sister were heterozygous for this novel mutation. Conclusion A novel TYMP gene mutation that caused MNGIE in a Chinese adult patient was confirmed by gene detection.
mitochondrial neurogastrointestinal encephalomyopathy; thymidine phosphorylase gene; gene mutation
吴 东 电话:010-69155014,E-mail:wudong061002@aliyun.com
R746
A
1674-9081(2014)03-0302-05
10.3969/j.issn.1674-9081.2014.03.011
2013- 08- 26)
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
国际医学放射学杂志(2021年4期)2021-08-05
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年3期)2021-07-22
医学新知(2019年4期)2020-01-02
农药科学与管理(2019年6期)2019-11-23
腹腔镜外科杂志(2016年12期)2016-06-01
中华老年多器官疾病杂志(2016年8期)2016-05-14
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
肿瘤预防与治疗(2015年5期)2015-09-26
