
基于免疫与神经网络的RNA二级结构预测*
2014-07-14 05:49李金铭
湖北科技学院学报 2014年7期
林 航,李金铭
(福建农林大学,福建 福州 350002)
脱氧核糖核酸具有重要的生物学功能,是DNA到蛋白质间遗传信息中间传递体[1]。RNA二级结构是特定平面图,配对的部分构成局部的双螺旋结构,这就是RNA的二级结构[2],它是一种平面的结构。目前,使用实验方法在RNA信息数据的惊人的增长下,实现生物分子的具体的二级结构有着其自身的局限性,而且对所有分子并不是都有效的[3]。
研究表明,对RNA二级结构预测,通过计算机仿真与数学建模的方法具有较高的可信度和参考价值,主要的类型有:
(1)动态规划的方法
动态规划方法是基于热力学原理。Zuker动态规划算法为代表的[4]的动态规划算法,是自由能计算模型。该算法实现简单,但它不能预测带假结的RNA分子,由于其真实结构可能不是最低自由能,所以用这种方法来预测准确率低。
(2)比较序列法
比较序列的方法通常采用的是共变模型[5]和随机上下文无关语法模型[6]。通过比较序列的方法可以预测RNA二级结构更好,更准确,但他们需要更多的同源序列,并且对同源序列还有一定的限制要求,花的时间也较多。
由于上述方法的局限性,组合优化算法被提了出来。通过使用这种方法,模拟退火算法、遗传算法[7-9]、Hopfield神经网络[10-11]等启发式算法被用来解决该问题。本文采用混沌Hopfield神经网络算法预测RNA二级结构。
一、混沌理论
混沌是一种非线性现象,广泛存在于自然界中,它可能看起来有些混乱,但其精致的结构,使随机性、遍历性和规律性成为它的特点,某个范围内,其可以按自身的规律不重复遍历所有状态。
一般指的是通过随机确定性方程获得的随机性运动状态,混沌状态变量被称为混沌变量。logistic映射是是一个常用的混沌系统,方程如下:
Zn+1=μzn(1-zn) n=0,1,…n
(1)
式(1)是一个动力学系统,μ作为其控制参数。μ值确定后,从任意的初值 z0∈[0,1],可以迭代得到一个确定的时间序列Z0,Z1,…Zn。 当μ=4 时,是一个完全处于混沌状态系统。混沌优化算法能够轻易的跳出局部最优解,归功于它的混沌性。它的搜索机制表现很好,常常被用于算法的混合中,并作了广泛研究。把混沌算子应用于遗传算法中,提出了混沌遗传算法,取得了较好的效果。把混沌搜索结合于微粒群算法,得到了混沌粒子群优化算法,都具有很好的搜索性能。
二、Hopfield神经网络
Hopfield神经网络是一个只有单一的神经元层次的结构,同时每个神经元与其他神经元的输出是相互连接的,称之单层反馈网络。0或者1两种值是Hopfield神经网络的取值集合。Yi(t+1)是神经元的输出,Xi(t+1)是网络的输出,如公式(1):
Xi(t+1)=sgn(yi(t+1),i∈[1,n]
(2)
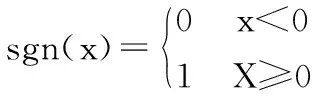
Hopfield神经网络在优化计算的应用中,目标函数有相应的能量函数,从而网络权值也被确定,当神经网络的能量达到最小时的解,就是问题的最优解。
三、混沌Hopfield神经网络优化算法求解
1.应用于RNA二级结构预测的Hopfield神经网络Hopfield神经网络用于RNA二级结构预测问题时,用基于最小自由能思想的茎区自由组合算法时,由于Vi=0表示该神经元被选中,能量函数可以写成[12]:
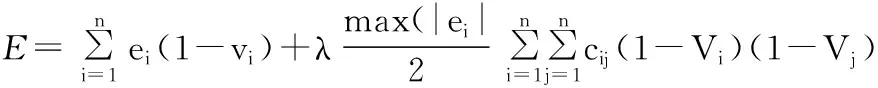
(3)
其中, cij即为节点i 和j 间的权值,茎是否被选中决定了Vi取0或1, ei表示茎i的能量;茎i 与j 是否相容来决定取值为0或1;非稳定茎和稳定茎间的相对率通过λ调整。
2.混沌Hofield神经网络算法描述
对于Hopfield神经网络来说,其对初始值的依赖性很强,因此本文使用距离函数产生初始解,对神经网络的初始解进行优化。对Hopfield初步产生的最优解,通过使用混沌函数对其值进行混沌化,使其跳出局部最优解,搜索全局最优解,其具体步骤如下:
(1) 定义问题,能量函数对应目标函数和约束条件,神经元的输出 Vi对应问题的解,初始化神经网络参数,令 T=max,t=0 ;
(2) 使用距离函数计算初始解,初始化神经网络;
(3) 初始化神经网络,选取一个还未被访问的聚类中心,为该神经网络神经元的初始输出;
(4) 当Ui>0 时,Vi=1 ,否则 Vi=0。而 Vi=0说明该碱基配对;
(5) 使用公式(4)中的定义的更新Ui,Vi;
(6) 当满足终止条件,即满足Ui(t+1)=Ui(t)+△Ui(t)△t或者△ Ui(t)或者t=max ,继续下一步, 否则转4);
(7) 计算当前解的目标函数值;
(8) 使用混沌函数对初步初始解进行混沌化,产生新的初始值初始化神经网络;
(9) 达到最大迭代次数,优化终止,从中选取使目标函数值最小的解。
四、实验结果
1. 评价标准
敏感性(X),特异性(Y)和马休兹相互作用系数 (MMC)是目前评价RNA二级结构准确率主要的3个度量参数。X是所有碱基对在真实结构中被正确预测到的比率。Y是正确预测所有预测到的碱基对的比率。一般折中衡量上述两个参数的是MCC。
2. 参数设置和仿真结果比较
仿真中,A=0.1,B=0.2 为神经挽留过的常量参数。最后得到实验结果见下表1。我们可以看出本文的算法优于其他算法。
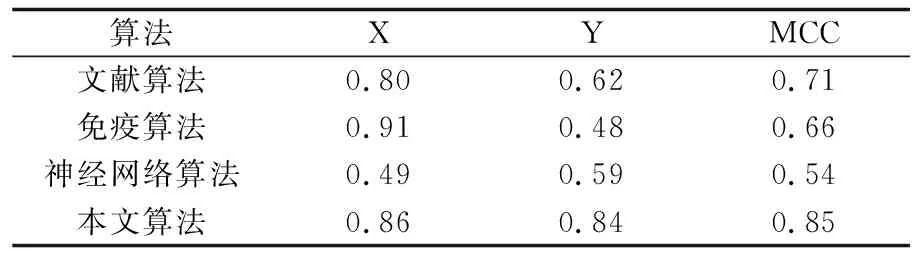
表1 4种算法平均预测准确率的比较
五、 结语
本文根据混沌算法的遍历性和随机性,和神经网络对初值的依赖性,结合RNA结构的保守性的特点,对神经网络的初值进行优化,将Hofield神经网络通过混沌函数进行优化,首次运用于RNA二级结构预测,提高了全局,搜索能力,使其不易陷入局部最优解,并获得一定的效果。
参考文献:
[1] 林娟,钟一文,张骏. 离散蛙跳算法预测RNA二级结构[J].南京师范大学学报(工程技术版), 2011,(4).
[2] 邢翀. RNA二级结构预测算法的研究[D].长春:吉林大学,2012.
[3] 林娟,钟一文. 改进的免疫粒子群优化算法预测RNA二级结构[J].计算机工程与应用, 2012,(1).
[4] ZUKER M. On finding all suboptimal foldings of an RNA molecular[J]. Science, 1989, 244(4900):48~52.
[5] Ennysr. Durbar RNA Sequence Analysis using covariance models[J].Nucleic Acids Res.1994,(22):2079~2088.
[6] Sakakbara Y,Browm M,Hugheryr.Recent methods for RNA modeling using stochastic context-free grammars[C]. Grochemorem.Gusfield D.Proceedings of the sikomar conference on combinatorial pattem matching Berlin:Springer-Verlag.1994:289~306.
[7] Cai L, Malmberg R. L, Wu Y. Stochastic modeling of RNA pseudoknotted structures: a grammatical approach [J]. Bioinformatics.2003:166~173.
[8] HU Yuh-Jyh. Prediction of consensus structural motifs in a family of coregulated RNA sequences [J]. Nucleic Acids Research , 2002 ,30(7) : 3886~3893.
[9] SHAPIRO B A , WU Jin-chu , BENGALI D , et al. The massively parallel genetic algorithm for RNA folding: MIMD implementation and population variation[J]. Bioinformatics , 2001 ,17 (2) : 137~148.
[10] WIESE K C , DESCHENES A A , HENDRIKS A G. Rna Predict-an evolutionary algorithm for RNA secondary structure prediction[J]. IEEE/ ACM Transactions on Computational Biology and Bioinformatics , 2008 ,(50) : 25~41.
[11] T AKEFUJI Y ,CHEN Li-lin , LEE K c, et al. Parallel algorithms for finding a near-maximum independent set of a circle graph[J]. IEEE Transaction on Neural Networks , 1990,1(3): 263~267.
[12]刘琦,张引,叶修梓. 基于离散Hopfield网络求解极大独立集的茎区选择算法以及在RNA二级结构预测中的应用[J]. 计算机学报,2008, 31(1): 51~58.
猜你喜欢
电子制作(2019年19期)2019-11-23
现代装饰(2018年5期)2018-05-26
中央民族大学学报(自然科学版)(2017年1期)2017-06-11
统计与决策(2017年2期)2017-03-20
重型机械(2016年1期)2016-03-01
现代计算机(2016年34期)2016-02-28
智能系统学报(2015年4期)2015-12-27
大连工业大学学报(2015年4期)2015-12-11
电源技术(2015年5期)2015-08-22
中国生化药物杂志(2015年4期)2015-07-07
