
脱氧胆酸对人食管腺癌细胞TNF-α、IL-8和 IL-6表达的影响及其机制
2014-06-27 12:04张蓉,单虎,刘欣,张军
西安交通大学学报(医学版) 2014年1期
张 蓉,单 虎,刘 欣,张 军
(西安交通大学医学院第二附属医院:1. 消化科;陕西西安 710004;2.心内科,陕西西安 710004)
近年来,流行病学资料显示,食管腺癌(esophageal adenocarcinoma, EAC)的发病率呈快速增加的趋势[1],尤其在美国白人中,EAC已经超越食管鳞癌成为食管癌最常见的组织学类型[2]。各种原因引起的酸和(或)胆酸等反流物反流至食管导致食管上皮慢性炎症,持续存在的炎症通过释放大量化学趋化因子、细胞因子及活性氧(reactive oxygen species, ROS)等,促使食管上皮细胞增殖并最终诱导细胞突变[3],这是目前普遍认同的EAC发病机制。
然而,单独抑酸治疗并不足以预防胃食管反流病进展为EAC[4]。因此,除酸外,胆酸在EAC的形成和进展中可能发挥重要作用。人体中的胆酸主要包括胆酸(CA)、鹅脱氧胆酸(CDCA)以及脱氧胆酸(deoxycholic acid, DCA),其中以DCA的毒性作用最强。DCA能引起食管上皮细胞活性下降及诱导慢性炎症发生[5],常作为肿瘤诱导剂用于EAC及其癌前病变的动物造模[6]。在EAC患者中,高浓度的胆酸通过释放ROS发挥其细胞毒性及遗传毒性作用,主要包括损伤细胞膜、线粒体膜及干扰细胞功能、造成DNA氧化损伤等[7],从而诱导肿瘤形成并参与肿瘤的进展,其次ROS也作为一个中间分子调节炎症诱导的肿瘤发生和发展[8]。然而,DCA对EAC炎症因子表达的影响以及可能的机制尚未见报道。本研究将探讨DCA对EAC细胞炎症因子表达的影响,分析DCA调节炎症因子表达是否通过ROS途径实现,为其发病机制的研究提供理论依据。
1 材料与方法
1.1材料人EAC OE19细胞株由第三军医大学西南医院消化科房殿春教授惠赠;RPMI-1640培养基(美国HyClone公司),胎牛血清(美国Gibco公司),重组人球形脂联素(美国Peprotech公司),重组人全长型脂联素及ELISA检测试剂盒(美国R&D Systems),Trizol和ROS检测试剂盒(美国Invitrogen公司),逆转录试剂盒(加拿大Fermentas公司),SYBR荧光染料检测试剂盒(中国TaKaRa公司),DCA(美国Sigma公司),NAC(中国Wolsen公司)。
1.2体外细胞培养将人EAC OE19细胞培养于含100 mL/L胎牛血清的RPMI-1640培养基及抗生素(100 U/mL青霉素及0.1 mg/mL链霉素)中,于37 ℃、50 mL/L CO2及950 mL/L湿度条件的培养箱内培养。用2.5 g/L胰蛋白酶消化传代,取对数生长期细胞用于实验,所有实验均重复3次。
1.3MTT分析细胞活性取对数生长期细胞,8×103细胞/孔接种于96孔板,细胞生长至培养瓶的80%,用无血清培养基继续培养24h,实验组DCA终浓度为50、100、150、200、250、300 μmol/L,空白对照组加入等体积的培养基,每浓度每时间点重复5孔,继续培养,分别于培养1、3、6、12、24 h加5 mg/mL MTT 20 μL,37 ℃孵育3 h直到结晶紫出现,弃上清液。每孔加二甲基亚砜(DMSO)150 μL,摇床振荡10 min,在492 nm波长酶标仪上测各孔吸光度(A)值。按公式:细胞活性=DCA组A值均数/对照组A值均数×100%。
1.4Real-timePCR(RT-PCR)检测TNF-α、IL-6和IL-8mRNA水平对数生长期细胞接种于6孔板,细胞生长至培养瓶的80%,用无血清培养基培养24 h,在有或无NAC存在的情况下,DCA(终浓度为50、100、200、300 μmol/L)及空白对照组(等体积的培养基)继续培养6 h。用Trizol试剂按说明提取细胞总RNA,取1 μg RNA,在20 μL反应体系逆转合成cDNA。用合适的引物及SYBR Green荧光染料扩增目的产物。TNF-α引物序列:上游引物5′-CCGTCTCCTACCAGACCAAGG-3′,下游引物5′-CTGGAAGACCCCTCCCAGATAG-3′;IL-8引物序列:上游引物5′-CCAAGGAGTGCTAAAGAACT-3′,下游引物5′-CTTCTCCACAACCCTCTG-3′;IL-6引物序列:上游引物5′-CACACAGACAGCCACTCACCTC-3′,下游引物5′-CTGCCAGTGCCTCTTTGCTG-3′;β-actin引物序列:上游引物5′-CCTGGGCATGGAGTCCTGTG-3′,下游引物5′-TCTTCATTGTGCTGGGTGCC-3′。引物均由北京奥科生物公司合成。每组设置1个复孔,每个基因重复3次试验。从扩增曲线获得Ct值,基因相对表达量=2-ΔΔCt,ΔΔCt=(Ct目的基因-Ct管家基因)实验组-(Ct目的基因-Ct管家基因)对照组。
1.5ELISA检测TNF-α、IL-6和IL-8蛋白表达细胞接种于6孔板,药物干预及培养时间同前。收集细胞培养上清液,4 ℃、3 000 r/min离心15 min,取上清液。用ELISA方法测定上清细胞因子TNF-α、IL-6和IL-8的水平,在450 nm波长酶标仪上检测样品吸光度(A)值,根据样品A值在标准曲线上查出相应TNF-α、IL-6和IL-8含量。
1.6流式细胞术测定细胞内ROS水平细胞接种于6孔板,在有或无NAC存在的情况下,采用不同浓度的DCA干预1 h后,去除细胞培养液,加入1 mL 10 μmol/L的荧光探针2′,7′-二氯二氢荧光素二乙酯(2′,7′-dichlorodihydrofluorescein diacetate, H2DCFDA),37 ℃培养箱内避光孵育30 min,用无血清培养液洗涤细胞3次。2.5 g/L胰酶消化收集细胞后,用500 μL PBS重悬细胞,1 h内上流式细胞仪检测双氯荧光素的荧光强度。
1.7统计学处理计量资料以均数±标准差表示,采用SPSS 17.0统计软件对各组数据进行正态性和方差齐性的条件检验;采用多因素的方差分析方法分析浓度和时间对OE19细胞活性作用及两者间的交互作用,采用单因素的方差分析方法分析不同浓度组细胞炎性细胞因子mRNA、蛋白水平以及ROS水平的差异,方差分析后两两比较采用LSD-t检验,P<0.05为差异有统计学意义。
2 结 果
2.1DCA对OE19细胞活性的影响DCA对EAC细胞活性的影响受到作用浓度和作用时间的共同影响(P<0.001和P=0.002),呈浓度依赖性及时间依赖性,随着作用浓度及时间的增加,细胞活性明显受到抑制(图1)。DCA干预1 h后,不同浓度的DCA对细胞活性无明显影响(P=0.067)。在3、12、24 h,低剂量的DCA(≤150 μmol/L)对细胞活性的影响与对照组之间的差异无统计学意义。但是,与对照组相比,高剂量的DCA(≥200 μmol/L)作用6、12、24 h后,细胞活性明显受到抑制(P<0.05),尤其是作用12、24 h,细胞活性明显下降了50%以上。此外,作用6 h后,高剂量DCA比低剂量DCA抑制细胞的活性更强(P<0.05)。因此,200 μmol/L的DCA作用6 h是其发挥生物学作用的初始浓度及有效时间。
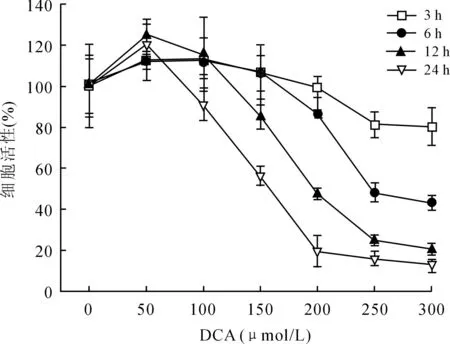
图1不同浓度DCA(μmol/L)对OE19细胞活性的影响
Fig.1 Effects of different doses of DCA (μmol/L) on viability of OE19 cells
2.2DCA对TNF-α、IL-6和IL-8mRNA水平的影响在OE19细胞中,DCA对TNF-α、IL-6 和IL-8 mRNA水平的影响是相似的,均呈现剂量依赖性(图2),与对照组相比,高剂量的DCA(200 μmol/L和300 μmol/L)明显增加TNF-α(6倍和9倍,P=0.001,P<0.001)、IL-8(6倍和10倍,P<0.001,P<0.001)和IL-6(3倍和6倍,P=0.001,P<0.001)。相反,DCA<100 μmol/L对TNF-α、IL-6和IL-8 mRNA水平的影响与对照组之间的差异无明显统计学意义(P=0.910,P=0.483,P=0.757)。与100 μmol/L DCA干预组相比,高剂量的DCA明显增加TNF-α、IL-6和IL-8 mRNA水平(P<0.05)。并且,NAC明显抑制DCA诱导的炎症因子TNF-α(1.9倍,P<0.001)、IL-8(2.2倍,P<0.001)和IL-6 mRNA水平(1.9倍,P=0.004)。
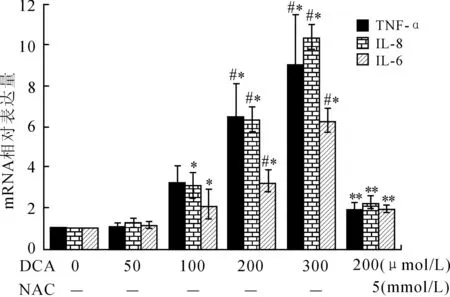
图2DCA与NAC对EAC细胞炎症因子mRNA的影响
Fig.2 Effects of DCA or/and NAC on the mRNA level of EAC inflammatory factors in OE19 cells
与对照组相比,*P<0.05;与低剂量DCA(≤100 μmol/L)相比,#P<0.05;与200 μmol/L DCA相比,**P<0.05。
2.3DCA对TNF-α、IL-6和IL-8蛋白水平的影响DCA明显增加TNF-α和IL-8蛋白表达,呈现剂量依赖性(表1)。低剂量的DCA(50 μmol/L和100 μmol/L)对IL-6蛋白的影响与对照组之间的差异无明显统计学意义(P=0.103,P=0.053),但是高剂量的DCA(≥200 μmol/L)明显增加IL-6的表达(P<0.001)。高剂量的DCA与低剂量的DCA相比,其TNF-α、IL-6和IL-8蛋白表达明显增强(P<0.001)。与DCA(200 μmol/L)干预组相比,NAC明显抑制其诱导的TNF-α、IL-6和IL-8蛋白的表达(P<0.001)。
表1DCA与NAC对EAC细胞炎症因子蛋白水平的影响
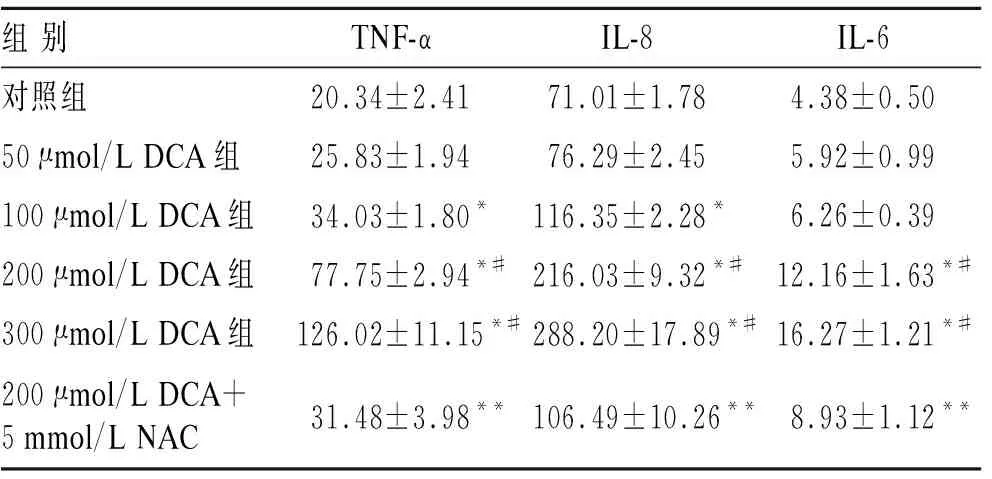
与对照组相比,*P<0.001;与低剂量DCA(≤100 μmol/L)相比,#P<0.001;与200 μmol/L DCA相比,**P<0.001。
2.4DCA对细胞内ROS的影响与对照组相比,随着DCA浓度增加,细胞内ROS水平明显升高,呈剂量依赖性(P<0.001,图3),并且高剂量的DCA(≥200 μmol/L)对细胞内ROS水平的影响比低剂量DCA(≤100 μmol/L)的作用更明显(P<0.001)。此外,ROS清除剂NAC明显减少DCA诱导的ROS水平(P<0.001)。

图3DCA(μmol/L)与NAC(mmol/L)对EAC细胞内ROS的影响
Fig.3 Effects of DCA (μmol/L) or/and NAC (mmol/L) on intra-cellular ROS level in OE19 cells
D1组:对照组;D2~ D5组:50、100、200、300 μmol/L DCA组;D6组:200 μmol/L DCA+5 mmol/L NAC。与对照组相比,*P<0.001;与低剂量DCA(≤100 μmol/L)相比,#P<0.001;与200 μmol/L DCA相比,**P<0.001。
3 讨 论
炎症与癌症的发生密切相关,其中有25%的实体瘤是由炎症引起的[9],其被认为是癌症形成的第7大危险因素[10]。各种化学损伤及应激事件通过触发细胞浸润及释放炎症细胞因子引发炎症反应及组织损伤。在炎症部位,活化的炎症细胞及免疫细胞能够产生ROS,引起DNA损伤、癌基因活化及抑癌基因失活促使肿瘤的形成和进展[8]。EAC是在胃食管反流物酸和胆酸作用下经过反流性食管炎-Barrett食管-异型增生等一系列过程进展而来,是胃食管反流长期作用形成的慢性炎症的并发症。胆酸反流至食管下段诱导食管癌变发生,也能够活化多种信号通路促使肿瘤细胞浸润和转移,其中尤以DCA的毒性作用最强。DCA通过诱发食管慢性炎症及食管损伤发挥其细胞毒性及遗传毒性作用,促使EAC形成和进展[8]。DCA的毒性作用与其浓度密切相关,胃食管反流患者体内DCA的浓度在0~280 μmol/L之间[11],高浓度DCA(>100 μmol/L)能诱导EAC细胞DNA损伤,相反,低浓度DCA(<100 μmol/L)无DNA损伤作用[5]。
本研究表明,低剂量的DCA(50~150 μmol/L)作用于OE19细胞对细胞活性无明显影响,高剂量的DCA(≥200 μmol/L)作用于OE19细胞后,随着药物浓度增加,作用时间延长,细胞活性明显受到抑制,呈时间-效应及剂量-效应依赖方式,说明高剂量的DCA具有明显的细胞毒性作用,亦是发挥生物学作用的有效浓度,这与SONG等[12]的研究结果相一致。因此,可以根据反流至食管下段的DCA的浓度将反流患者分为易患EAC的低危和高危人群,同时DCA的浓度也能作为一个分子标志物,用于预测反流患者罹患EAC的风险。
胆酸能诱导食管损伤及慢性炎症发生。慢性炎症主要通过改变细胞遗传学以及表观遗传学影响细胞周期,如DNA损伤和DNA甲基化或组蛋白修饰,诱导食管癌变。此外,持续存在的慢性炎症能够释放各种炎症调节物质,如ROS、活化的蛋白激酶、转录因子(CDX2、NF-κB)、炎症细胞因子及前列腺素等[13],促使细胞生长和浸润、细胞突变及血管形成,诱导肿瘤形成以及转移,其中ROS在炎症与食管癌变之间发挥着重要作用[8]。近来研究表明,EAC组织中促炎细胞因子NF-κB、COX-2、IL-1β、TNF-α、IL-8[14]和IL-6[15]明显高表达,并且酸和胆酸通过活化NF-κB促使EAC细胞表达IL-8和IL-1β[16]。DCA诱导的慢性炎症通过上调细胞内ROS水平促使EAC细胞DNA损伤以及NF-κB活化[5,7],ROS清除剂NAC以及抗氧化物质(维生素C)通过减少ROS释放能够抑制DCA诱导的DNA损伤[17]及NF-κB活化[5]。本研究发现,不同浓度DCA作用于OE19细胞后,与对照组相比,DCA明显上调炎症因子的表达及ROS水平,随着药物浓度增加,炎症细胞因子TNF-α、IL-8和IL-6 mRNA和蛋白水平及细胞内ROS水平均明显增加,呈剂量依赖性。采用ROS清除剂NAC与DCA联合作用后,能明显降低DCA诱导的ROS水平以及炎症因子表达。因此,DCA发挥促炎作用主要通过上调细胞内ROS水平实现。随着细胞内的ROS水平增加及机体的抗氧化能力下降,形成氧化应激状态,进而促使食管炎症形成,同时氧化应激也能活化一些转录因子(如NF-κB),促使基因转录,参与炎症反应及癌症的发生和发展[8,18]。
在EAC中,DCA具有明显的细胞毒性及促炎作用,呈剂量依赖性,DCA的促炎作用可能与细胞内的ROS水平密切相关。进一步研究产生ROS的来源以及寻找合适的抗氧化物质作为潜在的化疗药物用于EAC及癌前病变等相关疾病的治疗。
参考文献:
[1] SCHMASSMANN A, OLDENDORF MG, GEBBERS JO. Changing incidence of gastric and oesophageal cancer subtypes in central Switzerland between 1982 and 2007[J]. Eur J Epidemiol, 2009, 24(10):603-609.
[2] BROWN LM, DEVESA SS, CHOW WH. Incidence of adenocarcinoma of the esophagus among white Americans by sex, stage, and age[J]. J Natl Cancer Inst, 2008, 100(16):1184-1187.
[3] COUSSENS LM, WERB Z. Inflammation and cancer [J]. Nature, 2002, 420(6917):860-867.
[4] TRIADAFILOPOULOS G. Proton pump inhibitors for Barrett’s oesophagus[J]. Gut, 2000, 46(2):144-146.
[5] JENKINS GJ, CRONIN J, ALHAMDANI A, et al. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS[J]. Mutagenesis, 2008, 23(5):399-405.
[6] CHEN KH, MUKAISHO K, SUGIHARA H, et al. High animal-fat intake changes the bile-acid composition of bile juice and enhances the development of Barrett’s esophagus and esophageal adenocarcinoma in a rat duodenal-contents reflux model[J]. Cancer Sci, 2007, 98(11):1683-1688.
[7] JENKINS GJ, D’SOUZA FR, SUZEN SH, et al. Deoxycholic acid at neutral and acid pH, is genotoxic to oesophageal cells through the induction of ROS: The potential role of anti-oxidants in Barrett's oesophagus[J]. Carcinogenesis, 2007, 28(1):136-142.
[8] ABDEL-LATIF MM, DUGGAN S, REYNOLDS JV, et al. Inflammation and esophageal carcinogenesis[J]. Curr Opin Pharmacol, 2009, 9(4):396-404.
[9] HUSSAIN SP, HARRIS CC. Inflammation and cancer: an ancient link with novel potentials[J]. Int J Cancer, 2007, 121(11):2373-2380.
[10] COLOTTA F, ALLAVENA P, SICA A, et al. Cancer-related inflammation, the seventh hallmark of cancer:links to genetic instability[J]. Carcinogenesis, 2009, 30(7):1073-1081.
[11] NEHRA D, HOWELL P, WILLIAMS CP, et al. Toxic bile acids in gastro-oesophageal reflux disease: influence of gastric acidity[J]. Gut, 1999, 44(5): 598-602.
[12] SONG S, GUHA S, LIU K, et al. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett’s oesophagus and oesophageal adenocarcinoma[J]. Gut, 2007, 56(11):1512-1521.
[13] POEHLMANNA A, KUESTERA D, MALFERTHEINER P, et al. Inflammation and Barrett’s carcinogenesis[J]. Pathol Res Pract, 2012, 208(5): 269-280.
[14] COLLEYPRIEST BJ, WARD SG, TOSH D. How does inflammation cause Barrett’s metaplasia? [J]. Curr Opin Pharmacol, 2009, 9(6):721-726.
[15] DVORAKOVA K, PAYNE CM, RAMSEY L, et al. Increased expression and secretion of interleukin-6 in patients with Barrett’s esophagus[J]. Clin Cancer Res, 2004, 10(6):2020-2028.
[16] JENKINS GJ, HARRIES K, DOAK SH, et al. The bile acid deoxycholic acid (DCA) at neutral pH activates NF-kappaB and induces IL-8 expression in oesophageal cellsinvitro[J]. Carcinogenesis, 2004, 25(3):317-323.
[17] FOUNTOULAKIS A, MARTIN IG, WHITE KL, et al. Plasma and esophageal mucosal levels of vitamin C: role in the pathogenesis and neoplastic progression of Barrett's esophagus [J].Dig Dis Sci, 2004, 49(6):914-919.
[18] GRETEN FR, ECKMANN L, GRETEN TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer[J]. Cell, 2004, 118(3):285-296.
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
理化检验-化学分册(2021年10期)2021-11-29
中老年保健(2021年9期)2021-08-24
江西医药(2018年8期)2018-10-24
罕少疾病杂志(2017年2期)2017-02-23
中华胃食管反流病电子杂志(2017年3期)2017-01-16
畜牧兽医学报(2015年3期)2015-07-05
中国当代医药(2015年9期)2015-03-01
中成药(2014年4期)2014-04-01
