
连续催化在有机合成中的应用
2014-06-23 16:22:16段晓晓尹大学
合成技术及应用 2014年3期
张 严,费 莎,段晓晓,尹大学
(青海师范大学化学系,青海西宁 810008)
专题论述
连续催化在有机合成中的应用
张 严,费 莎,段晓晓,尹大学*
(青海师范大学化学系,青海西宁 810008)
总结连续催化法在有机合成常见反应类型中的研究进展和应用,例如取代反应、消除反应、氧化还原反应以及缩合反应等。分析比较连续催化工艺相对于传统的浓硫酸催化工艺的优点,并指出连续催化工艺在国内大规模工业生产中的不足之处。
连续催化法 取代反应 消除反应 氧化还原反应 缩合反应
催化剂是有机合成中常用的物质。目前,很多催化剂应用于某些有机合成反应时,经常出现催化剂单次使用的问题。不仅造成了催化剂的浪费,而且可能为后续产物的处理带来一些不必要的麻烦,使很多重要的有机合成反应只能局限于实验室中的小规模反应,不能适用于大规模的工业生产中。因此,提高催化剂的使用次数和保持催化剂的高效性以及稳定性是一项任重道远的工作。
连续催化,是一种在原有机合成反应原理的基础上,使用新型催化剂和新型反应装置于反应中,保证催化剂高效性和稳定性的同时,提高催化剂的使用次数,提高反应的转化率和产率,实现化学反应的可连续性操作,应用于大规模的工业生产。查阅大量国内文献发现,连续催化在国内有机合成中的应用也仅处在起步阶段,系统性介绍连续催化的文献较少。笔者对相关文献加以分类,总结出连续催化在有机合成中的应用现状。
1 连续催化在取代反应中的应用
取代反应是有机化学中最常见的一种反应类型。近年来,连续催化在取代反应中的应用日益增多。笔者根据不同的反应类型进行分类,主要分成连续催化酯化反应、连续催化水解反应、连续催化氨化反应以及连续催化烷基化反应等。
1.1 连续催化酯化反应
冯耀辉等[1]以NKC-9型阳离子交换树脂作为非均相固体酸催化剂,在填充固定床反应器中连续催化酯化废弃酸化油制备生物柴油。研究表明反应的最佳工艺条件:甲醇和油酸的摩尔比为2.8∶1,催化床层高为44.0 cm,进料流速为0.62 m L/min,反应温度为65℃。在该条件下,反应500 h,转化率始终保持在98.0%以上,并且NKC-9型阳离子交换树脂也未出现磺酸基流失的现象。生物柴油作为能源的一种替代物,早在20世纪80年代,上海内燃机研究所和贵州山地农机所承担此课题的研究,近些年,更加受到国内各大高校研究机构的重视。目前,海南正和生物制药有限公司,建成1 kt/a的生物柴油试验场,其技术和产品已于2002年10月通过国家经贸委新产品技术的鉴定;四川古杉油料厂已经建成生物柴油的生产装置,并投入运营。不久的将来,生物柴油可以更好地发挥能源替代物的作用。
盛梅等[2]以交联烯丙烯丙基葡聚糖凝胶(CADB)为载体,以对-β-硫酸酯乙砜基苯胺(SESA,结构式如图1)为偶联活性剂,共价固定工业脂肪酶,将油酸与十二醇发生酯化反应。实验结果表明:固定化酶采用间歇催化,每次24 h,重复使用30次,酯化率从93.5%降至47.0%;固定化填充在柱式固定床反应器中,采用连续催化,运行50 d,酯化率仍高达73%,提高了固定化酶的酶活率,继续改进,有望实现工业化大规模的生产。
霍稳周等[3]以丁二酸和甲醇为原料,采用自主开发的耐高温阳离子交换树脂为催化剂,采用无催化剂连续预酯化和连续催化精馏组合工艺合成丁二酸二甲酯。研究表明反应的最佳工艺条件:丁二酸和甲醇的摩尔比为1∶9,丁二酸的进料液态空速为0.7 h,反应温度为110℃。实验结果表明,耐高温阳离子交换树脂催化剂的稳定性很好,连续运行2 000 h,丁二酸的平均转化率为99.96%,丁二酸二甲酯的平均选择性为99.93%。
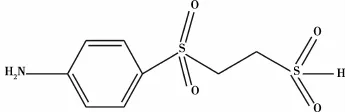
图1 对-β-硫酸酯乙砜基苯胺(SESA)的结构式
王金明等[4]使用负载双金属路易斯酸的树脂催化剂,采用连续催化工艺合成柠檬酸三乙酯(TEC,结构式如图2)。研究表明反应的最佳工艺条件:柠檬酸和乙醇的摩尔比为1∶3,空速1 m/(m ·h),反应压力1 MPa,反应温度为80℃,催化剂选用KC124树脂。实验结果表明,酯化反应的产率在90%以上,KC124树脂催化剂的稳定性好,其增塑的聚氯乙烯(PVC)可长期使用。
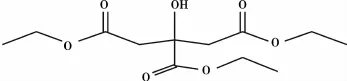
图2 柠檬酸三乙酯(TEC)的结构式
张云峰等[5]采用有机锡(β-甲氧羰乙基三氯化锡,结构式见图3)连续催化酯化合成乙酸乙酯,实验结果表明,此法比硫酸催化时的活性稍低,但使用寿命(115 h)更长,底物不碳化,催化剂可重复使用,且酯产率高于硫酸催化法;减少了使用浓硫酸作为催化剂导致的后续处理困难和对环境的危害,符合绿色化学的理念,有比较可观的应用前景。
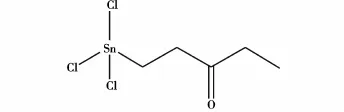
图3 β-甲氧羰乙基三氯化锡的结构式
黄科林等[6]采用GD001固体酸为催化剂,以乙酸与正丙醇为原料合成乙酸正丙酯,研究表明实验的最佳工艺条件:乙酸与正丙醇的摩尔比为1∶1.02,进料速度为150 m L/h(酸醇混合液),反应酸醇的摩尔比为3∶1~4∶1,反应温度为125℃。在该条件下,连续运行300 h后,催化剂仍具有很好的稳定性和寿命,酯化率达96%以上。
毛立新[7]采用硅胶固载硫酸钛在500℃下焙烧改性后的固体酸为催化剂,连续催化合成丙酸正丁酯。研究表明实验的最佳工艺条件:催化剂固载量0.2 g[Ti(SO4)2]/g(SiO2),丙酸和正丁醇的摩尔比为1∶1.4,进料量0.12 L/h。该条件下,丙酸正丁酯的收率在95.5%以上。
韦藤幼等[8]采用固体酸为催化剂,替代传统的硫酸催化剂,以醋酸和丁醇为原料合成醋酸丁酯。反应产生的粗酯经过精馏后,生产效率远远大于硫酸法,大大节省了投资和生产成本,粗产物中的醋酸丁酯的含量在96%以上,而且无副反应。
曾哲灵等[9]研究了浓硫酸连续催化樟树籽油酯化酯交换的实验。实验表明该工艺的最佳条件:甲醇和总脂肪酸(以癸酸和月桂酸为主)的摩尔比为5∶1,浓硫酸和总脂肪酸的摩尔比为0.6∶1,反应时间为7 h,反应温度为65℃。在该条件下,酯化反应酯交换率为91.12%。
孟祥河等[10]在无溶剂条件下,利用Lipozyme RM IM催化剂催化亚油酸和甘油连续合成1,3-甘油二酯(1,3-DAG,结构式见图4)。研究表明实验的最佳工艺条件:固定化酶填充柱长径比为7.8,亚油酸和甘油的摩尔比为1∶2,进料速度1.2 m L/min,反应温度65℃。在该条件下,过量的甘油可以吸附脱水来清除体系水分,并能明显改变固定化酶的稳定性,增加催化剂的使用寿命,连续运行10 d,残余酶活性保持在80%以上,而对照组仅为52%。
柯中炉等[11-12]采用强碱性阴离子交换树脂为催化剂,在固定床反应器中将菜籽油与甲醇进行酯交换反应,合成脂肪酸甲酯(FAME),进一步制备生物柴油。研究表明实验的最佳工艺条件:反应温度为60℃,醇油体积比为4.54∶1,反应时间为54.7 m in。在该条件下,FAME产品的收率为96.80%;反应温度为60℃,醇油体积比为5.96∶1,反应时间为54.7 min。生物柴油产品的收率为99.65%。实验结果表明:催化剂活性稳定,利用再生树脂连续催化28.7 h,未见其活性下降。
李连华等[13]利用固定床反应器,采用固定酸为催化剂催化桐油预酯化反应。实验优化的条件为:醇油摩尔比为6∶1,反应时间为88 m in,床层温度65℃。在该条件下,桐油的酸值可降低至0.8 mgKOH/g,达到生物柴油后续酯交换反应酸值小于1.0 mgKOH/g的要求。
于万鹏等[14]将加热至一定温度的邻苯二甲酸酐和甲醇,送入装有催化剂强阳离子交换树脂的串联式固定床连续催化试验装置,在催化剂的作用下连续反应生成邻苯二甲酸二甲酯。研究表明实验的最佳条件:甲醇和苯酐的摩尔比为2∶1,空速1 h-1,反应压力1 MPa,反应温度130℃,采用下进料方式,选用A45阳离子交换树脂,催化剂的含水量为0.5%以下。在该条件下,酯化反应产率可达90%以上,其增塑的PVC的力学性能接近邻苯二甲酸二辛酯(DOP,结构式见图5)和己二酸二辛酯(DOA,结构式见图6)增塑的PVC。传统的硫酸作为催化剂,虽然技术已经较为熟练,但存在设备腐蚀、不能连续生产、副产物较多、后处理较为复杂、所得产物色泽深、环境污染更为严重等问题。此法可以很好地解决上述问题,但工艺不够成熟,需要继续优化,将来有望改变目前采用浓硫酸为催化剂的主流工艺。方程式如图7所示。
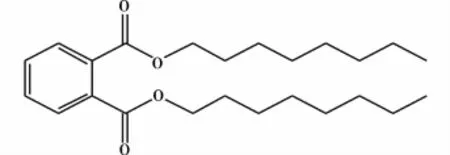
图5 DOP的结构式
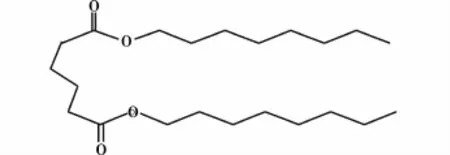
图6 DOA的结构式
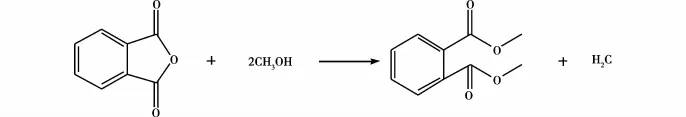
图7 合成邻苯二甲酸二甲酯的方程式
王存德等[15]制备一系列新型固体超强酸)并且利用其作为催化剂应用于各种酯化反应中。结果发现,固体超强酸不仅具有一般超强酸的性能,而且具有较高的催化活性和稳定性,连续催化活性基本保持不变。
刘红侠[16]研究了非均相体系中固定化脂肪酶(固定化假丝酵母脂肪酶)连续催化合成L-抗坏血酸棕榈酸酯(L-AP,结构式见图8)。研究表明实验的最佳条件:有机溶剂采取叔戊醇(20 m L),反应温度55℃,反应时间24 h,底物摩尔比为9∶1,底物浓度0.79 mol/L,固定化酶用量100U,体系水活度维持在0.1~0.3。实验结果表明:在该条件下,有利于L-AP的合成,产物浓度达14 g/L;流量为45 m L/min,即停留时间为2 min,更有利于L-AP的合成,加入分子筛(4A),对体系进行脱水处理,有利于L-AP的连续合成的进行,反应40 h后,L-AP浓度比未加分子筛的反应增加了约70%。
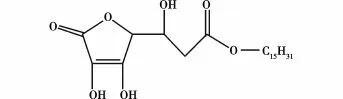
图8 L-AP的结构式
1.2 连续催化水解反应
颜涌捷等[17]采用FeCl3和FeCl2为催化剂,在液体连续流动的条件下,研究了木屑(主要是纤维素和半纤维素)在稀盐酸中的水解过程,转化成还原糖。实验结果表明FeCl2的催化效果比FeCl3显著,在适当的水解条件下,木屑的转化率可达71%以上。由于FeCl2比FeCl3的价格便宜,使用FeCl2作为催化剂不仅可以提高催化效果,还有利于减少反应的成本,投入到工业生产中,更好地产生经济效益。
目前,丙二酸的生产方法主要有:a)氯化镁氰化水解法,此法的缺点:氰离子有剧毒,对环境以及人的健康危害较大,反应过程较复杂;b)硫酸催化水解法,硫酸中的硫酸根不利于后续处理,并且容易与水中的钙离子结合,生成硫酸钙难溶物进入产品中,影响丙二酸的纯度;c)脂催化水解法,刘强等[18]以丙二酸二乙酯为原料,采用强酸性阳离子交换树脂为催化剂,通过水解反应制备丙二酸,研究表明实验的最佳条件:反应温度为80℃,丙二酸二乙酯和水的质量比为1∶3,流速4.5 m L/m in。,在该条件下,丙二酸的收率可达86.6%。此法具有以下优点:不易引进杂质,反应时间大大缩短,收率有所提高,有很好的发展前景。
1.3 连续催化氨化反应
邓信忠等[19]以CH2ClCH2Cl为原料,采用CuCl和2,2-联吡啶(结构式见图9)为催化剂,连续制备乙二胺。研究表明实验的最佳工艺条件为:反应温度120℃,反应压力4 MPa,氨烷比24.4∶1,催化剂加入量0.48 g,氨水质量分数60%,双催化剂质量比1∶1,反应停留时间7 m in。在该条件下,乙二胺的收率为64.24%,此法提高了产物收率,缩短了反应周期,降低了物耗,减少了HCl和NH3在空气中的逸散引起的环境污染,反应条件温和,安全性高。方程式如图10所示。
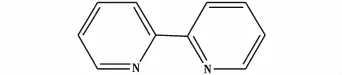
图9 2,2-联吡啶的结构式
1.4 连续催化烷基化反应
包燕芬等[20]探索采用白土系列-MS为催化剂合成邻位仲丁基酚的优化方法。研究表明实验的最佳工艺条件:反应温度(220±5)℃,苯酚和丁烯的摩尔比1∶0.6~0.7,体积空速700~800 h-1。方程式如图11所示。
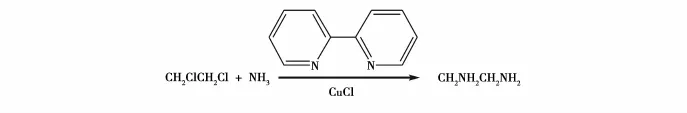
图10 合成乙二胺的方程式
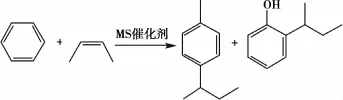
图11 合成邻位仲丁基酚的方程式
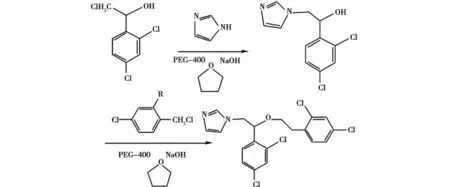
图12 合成硝酸咪康唑的方程式
王明慧等[21]将NaOH用作碱,采用PEG-400为相催化剂,在THF中不经分离连续完成1-(2,4-二氯苯基)-2-氯乙醇的N-和O-烷基化反应,制得抗真菌硝酸咪康唑和硝酸益康唑,收率分别为63%和 68%。本法操作简单,成本低廉,可以作为安全、高效的广谱抗真菌药,疗效好,有极高的发展应用前景,适合大批次工业化生产。方程式如图12-13所示。
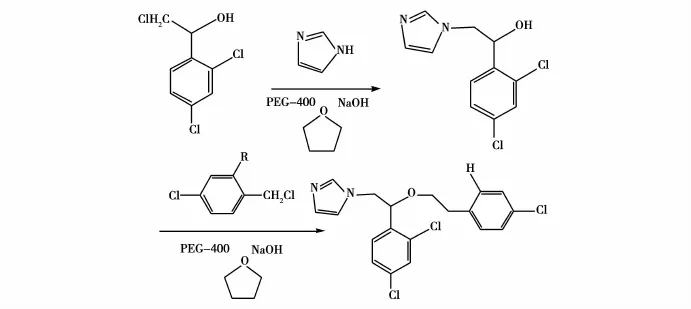
图13 合成硝酸益康唑的方程式
2 连续催化在消除反应中的应用
2.1 连续催化脱水反应
欧阳玉柱等[22]在直径为50 mm的催化剂反应精馏塔中,用活性炭固载硅钨酸作固定床进行合成环己烯的研究。实验结果表明,硅钨酸固定床催化剂精馏合成环己烯,催化活性高,选择性好,环境污染少,适合于工业生产,产物中烯含量可达99.5%。
2.2 连续催化脱氨反应
孙羽蒙等[23]在低压(<1.00 MPa)、氨气作为环境气体以及合适的催化剂,用一乙醇胺为原料,采用固定床连续催化合成哌嗪。研究表明实验的最佳工艺条件:反应温度256℃,反应压力0.90 MPa,氨气和一乙醇胺的摩尔比为5.63∶1。在该条件下,产物中哌嗪含率54.08%,产率25.15%。方程式如图14所示。
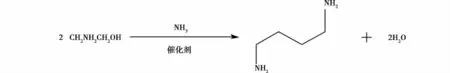
图14 合成哌嗪的方程式
3 连续催化在氧化还原反应中的应用
3.1 连续催化氧化反应
王昶等[24]在间歇操作的基础上,开发了甲苯连续催化氧化工艺。实验结果表明,当离心多孔气体分布器的转速为550 r/min,苯甲酸的收率可达50%。混合液体经过连续精馏不仅可得到苯甲酸,还可以得到附加值很高的苯甲醇、苯甲醛以及苯甲酸卞酯。
储振华等[25]采用CuCl2和MgCl2作为催化剂,采用连续的催化氧化法工艺,将2,3,6-三甲基苯酚氧化得到2,3,5-三甲基苯醌。实验结果表明,该工艺经过优化后,摩尔率可稳定在90%以上,产品纯度大于98.5%。方程式如图15所示。
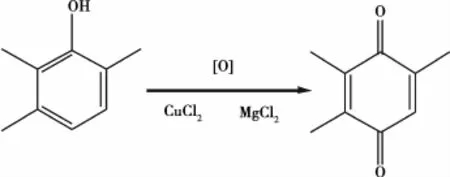
图15 合成2,3,5-三甲基苯醌的方程式
刘俊峰[26]以二甘醇为原料连续催化氧化合成1,4-二氧六环。研究表明实验的最佳工艺条件:反应温度240℃,催化剂的起始浓度:硅钼酸3%,或硫酸铁3%,或者硅钼酸银2%,连续补加浓度:硅钼酸1%,或硫酸铁1%,或者硅钼酸银0.5%。方程式如图16所示。以二乙醇为原料连续催化氧化合成1,4-二氧六环。研究表明实验的最佳工艺条件:反应温度190℃,催化剂的起始浓度:硅钼酸4%,或硫酸铁3.5%,或者硅钼酸银2%,连续补加浓度:硅钼酸1.5%,或硫酸铁1.5%,或者硅钼酸银1%。方程式如图17所示。
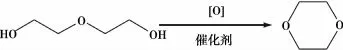
图16 二甘醇合成1,4-二氧六环的方程式
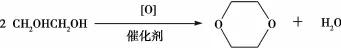
图17 乙二醇合成1,4-二氧六环的方程式
李长波等[27]以金属锰和铈的硝酸盐为活性组分的前驱体,以介孔分子筛SBA-15为载体,浸渍焙烧法制备了负载型MnO2/SBA-15、CeOX/SBA-15及MnOX-CeOX/SBA-15,并以H2O2为氧化剂,在温和条件下连续催化湿式过氧化(CWPO)处理腈纶废水。研究表明实验的最佳工艺条件:反应温度150℃,进料流量25 mL/min,催化剂投加30 g/L,H2O2投加5%。实验结果表明,MnOX-CeOX/SBA-15催化剂表现出较好的催化活性和稳定性,腈纶废水的COD去除率达到80%。
杭晓敏等[28]在实验室研究了3种连续环氧化反应工艺工业化运行的可行性。实验结果表明:离子交换树脂柱连续催化环氧化反应工艺和离子交换树脂柱与填料柱相结合的连续环氧化反应工艺无法成功运行,而采用多釜串联连续环氧化反应工艺在实验中取得成功。
张秋格等[29]在20 t/a连续放大装置上,以工业双戊烯(结构式见图18)为原料,采用Pd/C为催化剂,连续催化脱氢制备对伞花烃(结构见图19),实验结果表明,280℃是可选择和长期运行的反应温度,此时对伞花烃的产率为97.07%。

图18 双戊烯结构式
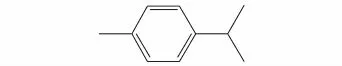
图19 对伞花烃结构式
吴强[30]对湿式氧化反应技术在连续处理废水方面的应用进行了研究。实验结果表明,采用活性炭及分子筛MCM-41为载体的铜催化剂的催化效率比较高,在苯酚、总有机碳(TOC)的去除方面均优于单一活性炭的催化效果,滴流床反应器能够在较低的反应温度(150℃)及压力(低于1 MPa)下,迅速有效去除90%以上的苯酚以及总有机碳(TOC)。
刘亚芹等[31]采用共沉淀法制备了钙钛矿型La0.8Sr0.2MnO3催化剂,并且考察了催化剂对氯苯的催化燃烧性能。研究表明实验的最佳工艺条件:氯苯浓度为800 mg/m3,空速(GHSV)为1 000 h-1,反应温度为410℃。在该条件下,经历了100 h连续催化燃烧后,催化剂没有出现明显的失活现象。
3.2 连续催化还原反应
杨军等[32]研究了双环戊二烯(DCPD,结构式见图20)连续催化加氢合成四氢双环戊二烯(THDCPD结构式见图21),筛选具有良好催化性能的Ni/γ-Al2O3催化剂。研究表明实验的最佳工艺条件:反应温度为150~160℃,氢气压力为1.0~1.2 MPa,空速为20~30 h-1。在该条件下,DCPD转化率高于98%,THDCPD收率可达95%以上。
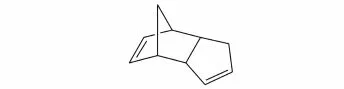
图20 双环戊二烯(DCPD)结构式

图21 四氢双环戊二烯(THDCPD)结构式
米镇涛等[33]以双环戊二烯(DCPD)为原料进行加氢,制备具有三环结构的四氢双环环戊二烯(THDCPD),以此为基础可制得一系列有价值的具有三环[5、2、1、02.6]癸烷的衍生物。研究表明实验的最佳工艺条件:选择高选择性APC-2催化剂,催化剂粒度在50~60目以下,采用正己烷为稀释溶剂,正己烷溶液内20%~30%为宜,反应温度160~180℃,反应压力1.0 MPa。
4 连续催化在缩合反应中的应用
4.1 连续催化羟醛缩合反应
王志宏[34]以甲醇和甲醛为原料,在硫酸催化下反应精馏合成甲缩醛。研究表明实验的最佳工艺条件:甲醇和甲醛的摩尔比为2∶1,精馏回流比8~10∶1,催化剂加入量为甲醇质量的15%。在该条件下,产品含量不小于98%,收率不小于91%。
4.2 连续催化酮醛缩合反应
刘天华等[35]以柠檬醛(结构式见图22)和丙酮为原料,采用大孔强碱性树脂(对假性树脂AX)作为催化剂,替代传统的NaOH作为催化剂,合成假性紫罗兰酮(结构式见图23)。研究表明实验的最佳工艺条件:反应液中柠檬醛/(丙酮+甲醇)=1∶5,其中丙酮/甲醇=4∶1,反应流速113 v/h,反应温度55℃。实验结果表明,AX催化剂经200 h的连续使用,再生后,重复使用性能较好,假性紫罗兰酮的产率为62.5%,收率为55.2%。
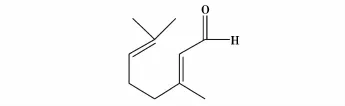
图22 柠檬醛的结构式
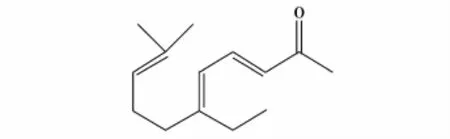
图23 假性紫罗兰酮的结构式
5 连续催化在酶催化反应中的应用
张传梅等[36]提出水凝胶载体材料优化模型,并制备出两种性能相异的载体材料,即聚丙烯酰胺/2-甲基丙烯酸羟基乙酯(结构式见图24)和聚N-异丙基丙烯酰胺(其单体结构式见图25)/2-甲基丙烯酸羟基乙酯,并在其中包埋α-胰凝乳蛋白酶,利用载体材料的温敏性可实现固定化酶活力的可控及连续可调,两种固定化酶连续催化8次,固定化酶活力并无明显下降。
周纪宁等[37]研究了固定化L-天冬酰胺酶连续净化血浆过程,并建立了动力学模型。
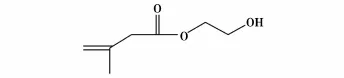
图24 2-甲基丙烯酸羟基乙酯的结构式
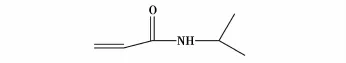
图25 N-异丙基丙烯酰胺的结构式
赵金爽[38]选择海藻酸钠为载体,固定化腈水合酶产生菌连续催化制备丙烯酰胺。研究表明实验的最佳工艺条件:流速为10 m L/m in,反应物丙烯腈的体积分数为3%,反应器装柱密度为6个/cm3。在该条件下,反应的固定化细胞酶的相对酶活性最高,可达94.2%,反应到6 h时,相对酶活性还可保持在60%左右。
6 结 语
综合文献,连续催化已经广泛地应用在各种不同类型的有机合成中,例如取代反应、消除反应、氧化还原等反应,特别是连续催化应用在取代反应中的实验较多,主要体现在合成酯以及制备生物柴油中。使用浓硫酸作为催化剂会带来的一些弊端,例如硫酸带来的环境污染;浓硫酸中的硫酸离子与水中的少量金属离子形成微溶物质,对产物质量的影响;浓硫酸催化法带来的复杂的后处理;不能使用于连续催化的问题等。连续催化法与一般的浓硫酸作催化剂相比较,一般是制备新型催化剂,增大催化剂的使用次数和寿命,从而减少催化剂的使用量;提高催化剂的高效性和稳定性,符合节能减排和绿色化学的理念,有很大的发展前景。但是,目前的文献报道中,鲜有连续催化的实验应用于大规模的工业化生产,其局限性主要有两点:第一,新型催化剂虽然比浓硫酸有很多可取之处,但连续催化法的工艺还不够成熟,很多新型催化剂的催化性能还需要较大的改善,方能满足投入工业生产中。另外新型催化能不能批次生产和生产的成本也是一个很重要的影响因素。第二,连续催化法需要更多的新型设备,目前文献中仅以反应床为主,尚不能达到工业化工艺的要求。总之,新型催化工艺具有连续性、高效性的优点,为有机合成提供了一个新方法;但是,新型催化剂和新型设备作为连续催化工艺的两个决定性因素,目前国内对其研究还不完备,仍需化学工作者的长期关注和不懈努力。
[1]冯耀辉,何本桥,李建新.阳离子交换树脂填充固定床连续催化酯化制备生物柴油[J].化工进展(增刊),2010,29:712~713.
[2]盛梅,曹国民,宋国强,郭登峰.固定化脂肪酶在有机相中催化酶化酯化反应[J].石油化工高等学校学报,1999,12(3):25~30.
[3]霍稳周,魏晓霞,田丹,等.连续催化精馏合成丁二酸二甲酯[J].石油化工,2013,42(2):181~184.
[4]王金明,董建国,毕晓红.连续催化法合成柠檬酸三乙酯增塑剂[J].塑料助剂,2012,2(总第92期):18~22.
[5]张云峰,张效霓,朱东升,等.有机锡连续法催化合成乙酸乙酯的研究[J].东北师大学报自然科学版,2001,33(1):122~124.
[6]黄科林,樊晓丹,黄焕生,等.连续催化酯化-精馏联合过程合成乙酸正丙酯的研究[J].安徽化工,2002,2(总第116期):16~18.
[7]毛立新.固载硫酸钛连续催化合成丙酸丁酯[J].化学工业与工程,2005,22(4):275~278.
[8]韦藤幼,童张法.连续催化反应精馏生产醋酸丁酯新工艺[J].化工设计,2003,13(3):14~16.
[9]曾哲灵,史亚亚,梁丽军,彭超.樟树籽油的浓硫酸连续催化酯化酯交换[J].南昌大学学报(理科版),2008,32(6):575~579.
[10]孟祥河,孙培龙,杨开,等.填充床反应器中酶法连续合成甘油二酯的研究[J].生物工程学报,2005,21(3):425~429.
[11]柯中炉,奚立民,朱魏.强碱性阴离子交换树脂催化菜籽油酯交换反应合成脂肪酸甲酯.离子交换与吸附,2010,26(2):145~152.
[12]柯中炉,奚立民,杨伟群,朱魏.基于强碱性阴离子交换树脂固定床催化制备生物柴油[J].中国粮油学报,2011,26(5):71~75.
[13]李连华,吕鹏梅,刘伟伟,等.固体酸连续催化桐油预酯化反应研究[J].中国油脂,2007,32(9):54~57.
[14]于万鹏,董建国,毕晓红.列管式固定床反应器连续催化合成邻苯二甲酸二甲酯的研究[J].塑料助剂,2012,4(总第94期):40~45.
[15]王存德,冯学兵.稀土固体超强酸催化合成酯的研究[J].石油化工,1994,23:166~169.
[16]刘洪侠.非水相体系中固定化脂肪酶催化合成L-抗坏血酸棕榈酸酯[D].北京:北京化工大学化学工程与技术,2010.
[17]颜涌捷,任铮伟.纤维素连续催化水解研究[J].太阳能学报,1999,20(1):55~58.
[18]刘强,张红梅,常宏宏,等.树脂连续催化制备丙二酸的工艺研究[J].太原理工大学学报,2004,35(5):565~567.
[19]邓信忠,裴世红,程怡,等.二氯乙烷连续氨化合成乙二胺[J].精细石油化工,2013,30(1):8~11.
[20]包燕芬,王亲宝.连续催化法合成邻位仲丁基酚条件的优化[J].辽宁化工,1999,28(3):180~181.
[21]王明慧,杨立荣,吴坚平.咪康唑和益康唑的PEG-400连续催化N-和O-烷基化反应制备[J].中国医药工业杂志,2005,36(8):458~459.
[22]欧阳玉柱,邹晓勇,彭清静,等.固体酸固定床催化精馏连续反应合成环己烯[J].吉首大学学报(自然科学版),2000,21(3):87~90.
[23]孙羽蒙.低压连续催化合成哌嗪[J].高校化学工程学报,1999,13(2):178~181.
[24]王昶,赵富贵,郝庆兰,等.苯甲酸工艺的节能减排以及连续化工艺的开发[J].广东化工,2012,39(2,总第226期):241~243.
[25]储振华,臧恒昌.连续催化氧化法制2,3,5-三甲基苯醌的可行性研究[J].广州化工,2011,39(9):104~151.
[26]刘俊峰.二甘醇或乙二醇连续催化合成1,4-二氧六环[J].精细化工,1996,13:52~54.
[27]李长波,赵国峥,张洪林.连续催化湿式过养化处理腈纶废水研究[J].水处理技术.2013,39(7):77~80.
[28]杭晓敏,雕鸿荪,裘爱咏,华聘聘.油脂连续环氧化反应工艺研究[J].中国粮油学报,1997,12(5):28~32.
[29]张秋格,毕良武,赵振东,等.工业双戊烯连续催化脱氢反应制备对伞花烃的20t/a放大实验[J].精细化工,2009,26(7):675~679.
[30]吴强.滴流床反应器湿式氧化连续处理废水的研究[D].浙江:浙江大学化学工程,2000.
[31]刘亚芹,蔡文祥,余中平,等.钙钛矿型La0.8Sr0.2MnO3催化剂对氯苯催化燃烧的性能研究[J].浙江工业大学,2007,35(5):477~481.
[32]杨军,郭建维,米镇涛,刘卅.双环戊二烯连续催化加氢[J].石油化工,1998,27(7):475~479.
[33]米镇涛,杨军,李德庆.双环戊二烯连续催化加氢的研究[J].化学通报,1997,(7):41~42.
[34]王志宏.连续催化合成甲缩醛工业研究[J].精细化工中间体,2012,42(2):38~40.
[35]刘天华,张新兵,高法银,周毅.大孔强碱树脂连续催化合成假性紫罗兰酮[J].江苏石油化工学院学报,1998,10(1):15~18.
[36]张传梅,付建伟,庄银风,毛陆原.温敏性水凝胶作为固定化酶载体的研究[J].河南科学,2006,24(5):683~686.
[37]周纪宁,金浩,江体乾.固定化L-天冬酰胺酶连续催化过程的数学描述[J].华东理工大学学报,1999.25(3):273~275.
[38]赵金爽.海藻酸钠固定化腈水合酶产生菌连续催化制备丙烯酰胺[D].黑龙江:哈尔滨理工大学应用化学,2011.
App lications of continuous catalytic m ethods in O rganic Synthesis
Zhang Yan,Fei Sha,Duan Xiaoxiao,Yin Daxue
(Department of Chem istry,Qinghai Normal University,Xining Qinghai 810008,China)
This article summarized the research progress and app lications of continuous catalytic method in common organic synthesis reactions,such as substitution reactions,elimination reactions,redox reactions etc.By comparing with the traditional sulfuric acid catalytic process,we analyzed the advantages of continuous catalytic method and pointed out the disadvantages continuous catalytic method in domestic large-scale industrial production.
Continuous Catalytic method;substitution reaction;elim ination reaction;redox reaction;condensation reaction
TE624.4
A
1006-334X(2014)03-0028-08
2014-05-23
国家自然基金资助项目(21262025)
张严(1988—),男,安徽蒙城人,在读硕士研究生,研究方向为绿色有机合成。
*通讯作者:尹大学(1964—),男,博士,主要从事化学、化工的教学与研究工作。
猜你喜欢
宁夏医学杂志(2020年4期)2020-03-01 13:16:20
宁夏医学杂志(2020年3期)2020-02-27 14:17:11
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
纺织科学研究(2017年6期)2017-07-03 12:14:14
纺织科学研究(2017年1期)2017-05-17 03:59:17
中学生数理化·高三版(2016年12期)2017-03-02 19:21:37
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中国资源综合利用(2016年4期)2016-01-22 08:27:23
