
水合Pb(OH)+在高岭石(001)晶面的吸附机理
2014-06-23 06:51夏树伟于良民
物理化学学报 2014年5期
王 娟 夏树伟 于良民
(1中国海洋大学化学化工学院,海洋化学理论与工程技术教育部重点实验室,山东青岛266100;2青岛农业大学化学与药学院,山东青岛266109)
1 Introduction
The heavy metal elements,as one of the important pollutants in water environment,have attracted global concern due to their nonbiodegradable and persistent nature.1,2The particular toxicity of lead has been well established by the Agency for Toxic Substances and Disease Registry of the U.S.Department of Health and Human Services3since the year of 1999.Pb(II)is the common oxidation state of lead under normal environmental conditions,and hydrolysis of Pb(II)often occurs at slightly alkaline water environment,where Pb(OH)+is one of the most important species.Despite of the abundant biochemical data about Pb(II),much less effort has been devoted to understanding the fundamental aqueous chemistry of Pb(OH)+as well as its adsorption behavior on the mineral surface,especially from the molecular or atomic level.
Kaolinite,one of the plentiful natural clays,is a 1:1 layered aluminosilicate mineral with a unit cell composition of Al4Si4O10(OH)8.It has been widely used as the adsorbent for the removal of heavy metals from contaminated groundwater4,5due to its high specific surface area,good chemical,and mechanical stability,etc.The removal process for heavy metals takes place either through ion exchange on the permanent negative charge sites usually caused by isomorphic replacement of Si4+by Al3+in the silica tetrahedrons,6or through adsorption on the pH-dependent variable charge sites(S(OH))of the alumina face and the crystal edges,7or a combination of both.
Although a variety of batch tests about Pb(II)uptake by kaolinite have been reported8,9with the hydrolysis of Pb(II)considered,the developed empirical or semiempirical surface complexation models are only applicable for the specific experimental conditions,and sometimes they even don't fit to the data well.Also,the individual behaviors of Pb2+and Pb(OH)+can not be distinguished,including the preferred complexation mode,adsorption position,etc.Moreover,Pb(II)usually appears as an intermediate acid(with respect to the hard and soft acids and bases(HSAB)theory10),able to bind wide families of ligands in very flexible coordination modes11with hemi-directed or holo-directed coordination geometry.12These versatile characters of Pb(II)have further made the adsorption behavior of Pb(OH)+on kaolinite surface complicated.
Quantum chemistry calculations,especially the density functional theory(DFT)investigations,have become popular to complement experimental data recently.Although a large amount of DFT investigations on the structural and electronic properties of Pb(II)compounds have been carried out,13theoretical model about Pb(OH)+adsorption on kaolinite surface inclusive of the species and geometry of complex has not been found yet.
A first-principles density functional study of Pb(OH)+adsorption on the basal octahedral Al(Al(o))(001)surface of kaolinite was performed in this work.The water environment was considered.The monodentate and bidentate complexes,coordination geometry,and coordination number(CN)of Pb(II)as well as the two types of Al(OH)sites were examined.Feasibilities of different types of complexes were evaluated by the values of corresponding binding energies and the similarity of structure parameters to the experimental data.The related charge population and density of states(DOS)were also analyzed in detail to investigate the Pb―O bonding mechanism.
2 Theoretical methods and models
2.1 Computational methods
All calculations were performed with plane-wave pseudopotential DFT method utilizing the Cambridge Sequential Total Energy Package(CASTEP)code14in Materials Studio 5.0.Structures of the kaolinite bulk and(001)surface models were optimized with Perdew-Burke-Ernzerhof(PBE)15functional of the generalized gradient approximation(GGA),as it could describe molecular bonding(including hydrogen bond strength16,17)to greater accuracy than the local density approximation(LDA).Vanderbilt ultrasoft pseudopotential18in conjunction with a cutoff energy of 380 eV was adopted throughout.Monkhorst-Pack meshes ofk-point sampling19were generated with(4×2×3)and(2×2×1)grids for the kaolinite bulk and surface models,respectively.Higher cutoff energy or more refinedkpoint mesh only caused neglected changes in the results.Atomic positions were optimized by Broyden-Fletcher-Goldfarb-Shanno(BFGS)scheme with Gaussian smearing width of 0.20 eV until the total energy changed,the maximum tolerances of the force,and displacement converged to less than 1.0×10-5eV·atom-1,0.3 eV·nm-1,and 1.0×10-4nm,respectively.
Optimization of kaolinite bulk with chemical composition of Al4Si4O10(OH)8was started from the experimental structure reported by Bish.20The resulted unit cell parameters werea=0.5212 nm,b=0.9052 nm,c=0.7506 nm andα=91.813°,β=105.001°,γ=89.778°,within the error of 1.6%to the experimental values.Similar results have also been obtained by Hu and Michaelides21based on their DFT calculations.Structures of kaolinite and the(001)slab are shown in Fig.1 from a side view.Different from the kaolinite bulk,inner surface hydroxyls in the(001)slab could be clarified into two types:one third“lying”hydroxyls(OlH)oriented parallel to the kaolinite(001)surface and two thirds“upright”hydroxyls(OuH)oriented perpendicularly to the surface.The orientation of inner hydroxylsin the slab also changed,not parallel to the(001)surface as in the kaolinite bulk.
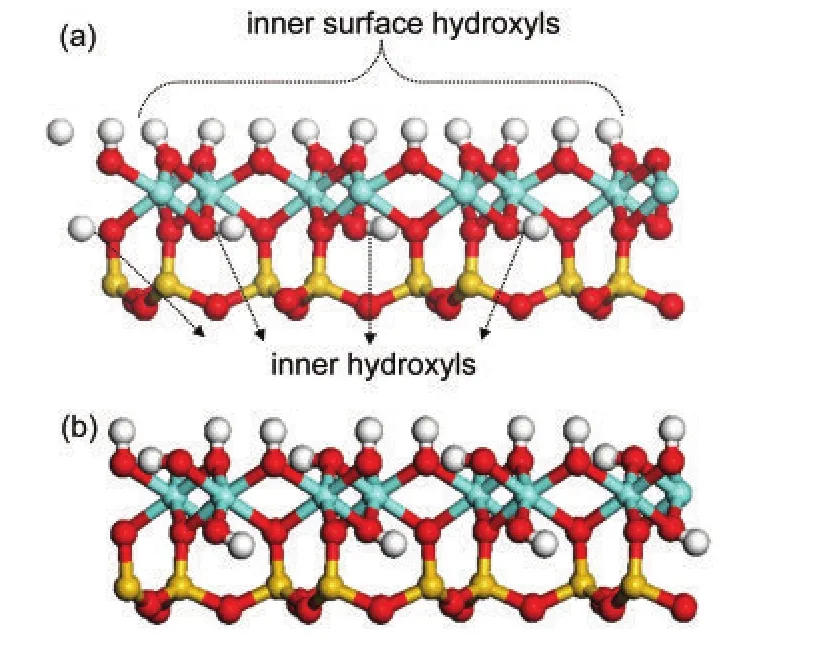
Fig.1 Side view of kaolinite structure with hydroxyl positions indicated(a)and the optimized geometry of(001)slab from the same visual point(b)
As aqueous chemistry of Pb(OH)+in solution was not well known,all possible species ofwere tested in a 1.0 nm×1.0 nm×1.0 nm periodic box with GGAPBE,ultrasoft pseudopotential,a cutoff energy of 380 eV,and 1×1×1k-point sampling.The same tolerances for self-consistence with total energy of 1.0×10-5eV·atom-1,force of 0.3 eV·nm-1,and displacement of 1.0×10-4nm were adopted to be consistent with the substrate optimization.
2.2 Surface models
The kaolinite(001)slab of a single two-sheet layer can be clarified into“H-O-Al-O-Si-O”six atomic“sublayers”.Based on the comparison of calculations on one-layer to two-layer models and one-layer models with different numbers of relaxed and fixed“sublayers”,a(2×1×1)surface unit cell(1.046 nm×0.907 nm×2.030 nm)was adopted as the substrate for Pb(OH)+adsorption,where the top four“sublayers”were relaxed and the bottom two“sublayers”were fixed during the optimization.The same atomic constraint has been used for uranyl adsorption study on(001)surfaces of kaolinite by Kremlevaet al.22The slabs were repeated periodically in thezdirection with a vacuum gap of 1.5 nm.Surface models with Pb(OH)+adsorbed were optimized under the same conditions to the clean(001)slab.
As shown in Fig.2,four types of surface complexation are explored.Monodentate complex of Pb(OH)+on Al(o)surface might form on the“Ou”site with“up”hydrogen or“Ol”site with“lying”hydrogen of any Al center since they belong to different types of inner surface hydroxyls,corresponding to configuration A or B,respectively.As the joint coordination of Pb(II)and H to the same surface oxygen center is possible,23bidentate complexes located on“OuOl”site of single Al center(C)and two neighboring Al centers(D)are also tested,where only the joint coordination structure of“Pb―Ol―H”is considered.
The binding energy(ΔEbind),calculated according to the reaction(1)based on the deprotonation of surface hydroxyl groups,is defined as the energy difference between the products and reactants.
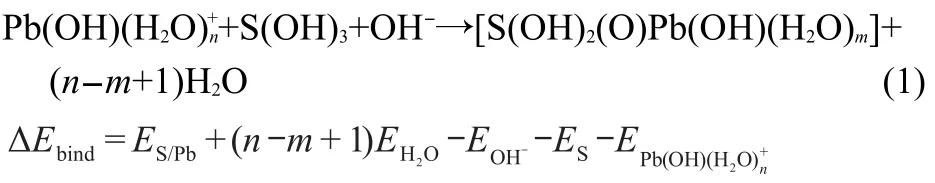
where S stands for the surface of the substrate,nandmare the numbers of H2O ligands in aqueous Pb(OH)+and the adsorption complex,respectively,ESandES/Pbrepresent the total energies of clean kaolinite(001)slab and the slab with Pb(OH)+adsorbed on the surface,respectively.“OH-”acts as an example group in alkaline aqueous system,which can react with the missed“H+”of S(OH)3here.
Bond length of Pb―O is taking as the main criteria to evaluate the reasonability of complex structure.In four types of adsorption modes,all possible species of[S(OH)2(O)Pb(OH)(H2O)m]withmvarying from 1 to 7 are tested.Complex from the DFT optimization having moderate Pb―O length of less than 0.37 nm24will be retained,otherwise be ruled out.As the water environment is considered in our work,Pb(II)tends to combine aqua ligands as many as possible.So only reasonable complex with maximum value ofmis adopted and analyzed.
3 Results and discussion
3.1 Geometries of Pb(OH)(H2O)n+
Pb(II)has a[Xe]4f145d106s26p0electronic configuration,where the“stereochemically active lone pair of electrons”of 6s2can take up more space on a specific region of the surface of coordination sphere than a single bond does,12and gives rise to the uneven distribution of ligands around Pb(II)ultimately.During the periodic DFT optimizations,one or more aqua ligands of Pb(OH)(H2O)+6-8left the first hydration shell withRPb―O>0.37 nmto reach the solvation sphere,indicating that the maximum value ofnwas 5 here.As for the geometries of Pb(OH)shown in Fig.3,complexes of Pb(OH)exhibited hemidirected geometry with increasing tendency to the holo-directed structure,while geometry with one“upright”water molecule occupying the other hemisphere of Pb(II)coordination globe was observed for Pb(OH)Combined with our previous findings that aqueous Pb(II)mainly exists in the structure of PbPb(OH)may be the most probable species of aqueous Pb(OH)+and is used as the initial adsorbate of kaolinite throughout.
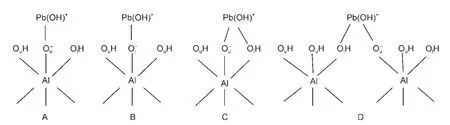
Fig.2 Schematic representation of the monodentate complexes on“Ou”(A)and“Ol”(B)sites and bidentate complexes on“OuOl”site of singleAl center(C)and two neighboringAl centers(D)
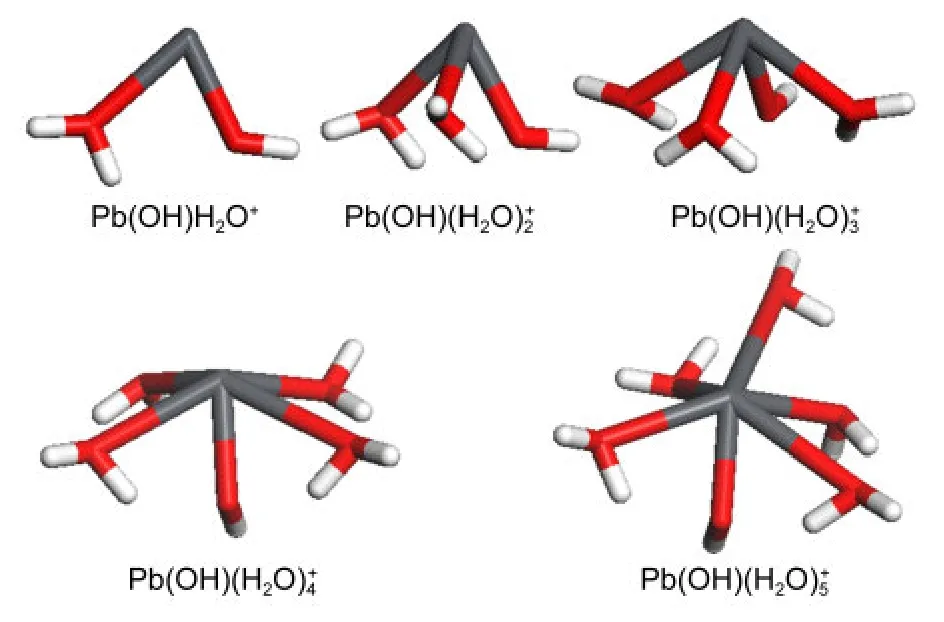
Fig.3 Equilibrium geometries of Pb(OH)(H2O)from periodic DFT calculations
3.2 Adsorption complexes
3.2.1 Monodentate complexes
In the monodentate adsorption mode,Pb(OH)+combines with the deprotonated substrate of[S(OH)2O]-,forming the overall neutral surface complex species.Withmranging from 1 to 7,all possible monodentate complexes of[S(OH)2(O)Pb(OH)(H2O)m]were tested.Unreasonable structures of“Ou”site with much longer Pb―O bond length were obtained until CN of Pb(II)was lower than 6,corresponding to the maximummvalue of 3(shown in Fig.4A).Two water molecules of Pb(II)hydration shell in aqueous system were crowded out by the kaolinite(001)slab due to the steric hindrance effect.Furthermore,stability and CN of complex were found to depend strongly on the hydrogen bonding interactions between aqua ligands and surface hydroxyls,especially the strong attraction of surface Olto H of aqua ligands(forming the“Ol…Hw”bond).Hydrogen bond between O of aqua(or hydroxyl)ligands and H of“Ou”site(designated as“Ow…Hu”or“OH…Hu”)also occurred,but was relatively weak and was less of a factor for the stability of complex.
As for the“Ol”site,only one water molecule could retain in the coordination sphere of Pb(II),corresponding to the maximum CN of 3(Fig.4B).Although atom of“Ol”exhibited stronger affinity to Pb(II)than“Ou”with a shorter Pb―Olbond length of 0.215 nm(Table 1),repulsive interactions from six surrounding“OuH”groups drove most of the aqua ligands of Pb(OH)away.No hydrogen bond of“Ol…Hw”formed as the distance between neighboring Olcenters was about 0.52 nm,too far apart for Hwto interact directly.
Both monodentate complexes were in hemi-directed geometry,where bond lengths of Pb―Osand Pb―OHwere obviously shorter than Pb―Ow.An even distribution of hydroxyl and aqua ligands just above the basal Al(o)(001)surface in configuration A has been found,associated with Pb-Al distance of 0.361 nm and three strong“Ol…Hw”bonds at distances of0.167,0.159,and 0.166 nm,respectively(Table 1).The Pb―Oubond length of 0.224 nm was slightly lower than the corresponding EXAFS(extensive X-ray absorption fine structure)value of(0.230±0.001)nm in the Pb(II)complex on kaolinite surface,26but in consistent with the Pb―O length of 0.219-0.232 nm for Pb(II)complex on aluminum oxides from the EXAFS spectroscopy.27Being different,configuration B displayed the distorted trigonal pyramid geometry with Pb―Ollength of 0.215 nm,Pb―Al distance of 0.313 nm,and the only one Pb―Owbond of 0.296 nm.
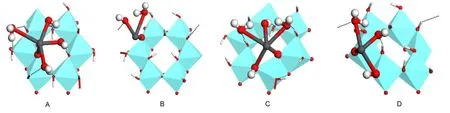
Fig.4 Equilibrium geometries of monodentate complexes on“Ou”(A)and“Ol”(B)sites and bidentate complexes on“OuOl”site of the sameAl center(C)and two neighboringAl centers(D)
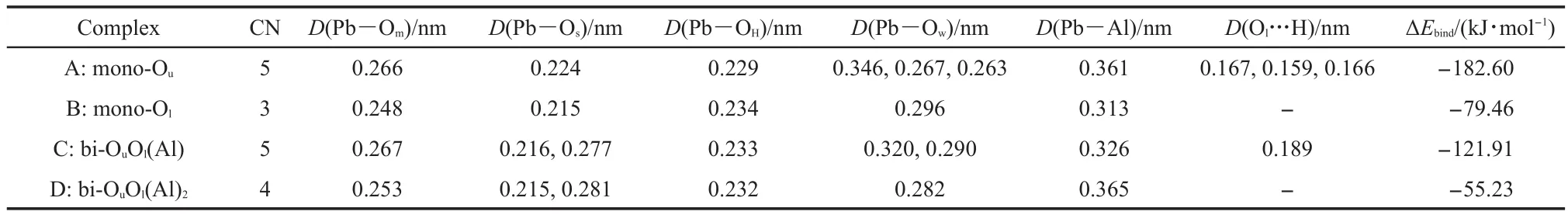
Table 1 Structure parameters and binding energies(ΔEbind)of monodentate and bidentate complexes of Pb(OH)+on theAl(o)(001)surface of kaolinite
Binding energies calculated from reaction(1)were-182.60 and-79.46 kJ·mol-1(Table 1)for configurations A and B,respectively.The negative values indicated the exothermic and favorable characteristic of the Pb(OH)+adsorption process.Thus,complex of“Ou”site seems more favorable based on the high binding energy,strong hydrogen bonding interactions,and good agreement of Pb―Oubond length with experimental values as well.
3.2.2 Bidentate complexes
Bidentate complexes with joint coordination structure located on single Al and two neighboring Al centers could be seen from Fig.4C and 4D,respectively.Both complexes featured hemidirected geometry with short Pb―Oubonds(0.215 and 0.216 nm)and slightly long Pb―Olbonds(0.277 and 0.281 nm).The weak Pb―Olbond was mainly caused by the competition of H with Pb to the Olcenter.All hydroxyl and aqua ligands of Pb(II)formed hydrogen bonds with surface hydroxyls,where the“Ol…Hw”type was stronger than“Ow…Hu”or“OH…Hu”.Bond lengths of Pb―OH(0.233 and 0.232 nm)were comparable to the corresponding value in Pb(OH)revealing that the combination of Pb(II)with Al(o)surface did not affect the bond of Pb―OHsignificantly.The main difference was that at most two water molecules could remain in the Pb(II)coordination sphere for configuration C,whereas only one aqua ligand left in configuration D.In other words,the maximum CN of Pb(II)was 5 for bidentate complex of single Al center,and 4 for that of two neighboringAl centers.Inspiringly,the Pb-Al distance of 0.326 nm in configuration C was in good agreement with the corresponding EXAFS data of 0.316-0.332 nm in the Pb(II)complex on aluminum oxides27and 0.327-0.336 nm for Pb(II)complex on iron oxides.28Combined with the binding energies of-121.91 and-55.23 kJ·mol-1for respectively configurations C and D,complexation in bidentate way seems less likely to occur on two neighboring Al centers,where the distance between“Ou”and“Ol”might be too large for Pb(II)to interact with both the atoms simultaneously.
Analysis focused on the two preferred adsorption types:monodentate complexation of“Ou”site(A)and bidentate complexation of single Al center(C).Configuration A featured a relatively high binding energy of-182.60 kJ·mol-1,incorporating the adsorption energy of Pb(OH)+,dissociation energies of two aqua ligands from Pb(OH)and hydration energy of“H+”from deprotonation of surface hydroxyl group.Although value of ΔEbind(-121.91 kJ·mol-1)for configuration C was slightly low,dissociation energy of one more water molecule from Pb(OH)was incorporated as the complex has a CN of 4,lower than that of complex A.Thus,the adsorption energies of Pb(OH)+in two types might be comparable.Moreover,both the Pb―O bond length of 0.224 nm in configuration A and Pb-Al distance of 0.326 nm in configuration C were in qualitative agreement with the EXAFS data.So these two types of adsorption were all probable,with monodentate complexation more easily to occur.
3.3 Properties
3.3.1 Mulliken population
Mulliken atomic populations of atoms Ou,Ol,OH,and Pb before and after the adsorption are listed in Table 2,which helps to quantify the charge transfer induced by Pb(OH)+adsorption.Charges for Ouand Olin clean kaolinite slab without adsorbent were respectively-1.06 and-1.05,indicating the comparable electronegativity of two different types of surface oxygens.Charges for OHand Pb in Pb(OH)before the adsorption were-0.98 and+1.26,respectively.Upon values of charge difference(Δ),Pb accepted about 0.20 electrons from the surrounding oxygen ligands during the adsorption process,including Ou,Ol,OH,and Ow.Therefore,covalency of Pb has increased after it combined with theAl(o)(001)surface.
Bond population of Pb―Oshas also been calculated to investigate the corresponding bonding mechanism(Table 2).Bonds of Pb―Ou,Pb―Olin monodentate complexes and Pb―Ouin bidentate complexes all featured negative population values,suggesting that filled antibonding interaction was the major orbital contribution.Positive values of Pb―Olpopulation in the joint coordination structure have been found,indicating the predominantly bonding orbital interactions.As values of Pb―Olpopulation were small,the formed Pb―Olbonds were very weak with notably long bond lengths of about 0.280 nm.So it seems that while hydrogen and Pb(II)coordinate to the samesurface Olsimultaneously,the coordination between hydrogen and Olis stonger than that of Pb(II).
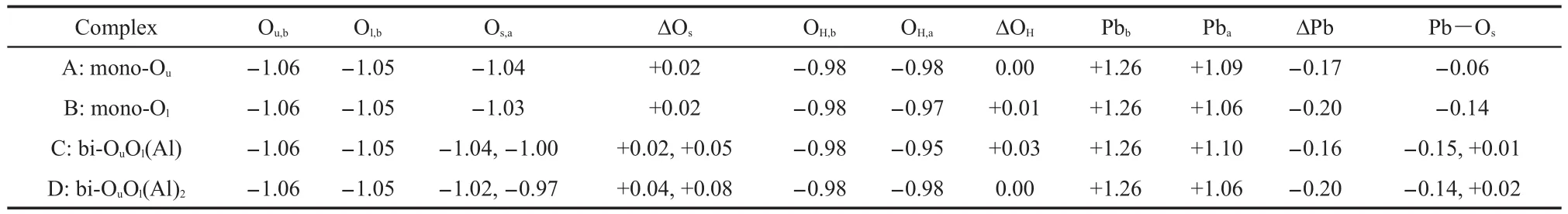
Table 2 Mulliken charges for atoms of Os,OH,Pb,and Pb―Osbond population in different types of adsorption complexes
3.3.2 DOS analysis
Further insight into the bonding nature of Pb(II)with hydroxyls of Al(o)(001)surface could be examined by the partial electronic densities of states(PDOS)of Ou,Oland Pb in four types of adsorption complexes.As shown in Fig.5A,combination of Pb(II)with Ouaffected the Ou2ssignificantly,as it has split from one peak(a)into two narrow parts(b,c,and d),with the corresponding energy shifted from-20.0--17.0 eV to a relatively high region of about-20.0--14.0 eV.Peaks in dashed curve at valence band region of-8.0--6.0 eV(a),as part of Ou2pcontribution in the clean Al(o)surface,decreased from the highest value of~1.10 electrons·eV-1(a)to ~0.20 electrons·eV-1(b,c,and d),while those at the conduction band of 4.0-7.0 eV increased from the highest value of~0.02 electrons·eV-1to ~0.20 electrons·eV-1after the adsorption.These changes are mainly caused by the Pb-Ouinteraction,Pb 6pcoupling with the Pb 6s―Ou2pantibonding states.As Pb 6sand Ou2pstates are located in the similar energy region,an efficient interaction that produces strongly antibonding Pb 6s―Ou2pstates occurs mainly at-5.0-0.5 eV.Coupling of Pb 6pwith the antibonding states can be obviously known from the overlaps between Pb 6s,Pb 6p,and Ou2pat-5.0-0.5 eV.Antibonding orbital interaction in chemical bonding is a characteristic of Pb(II)compounds.Similar interaction has been found in PbO29and hydrated Pb(II).25Also,it has been reported that the minimization of these unfavourable covalent antibonding interactions is the driving force for structural distortion.30
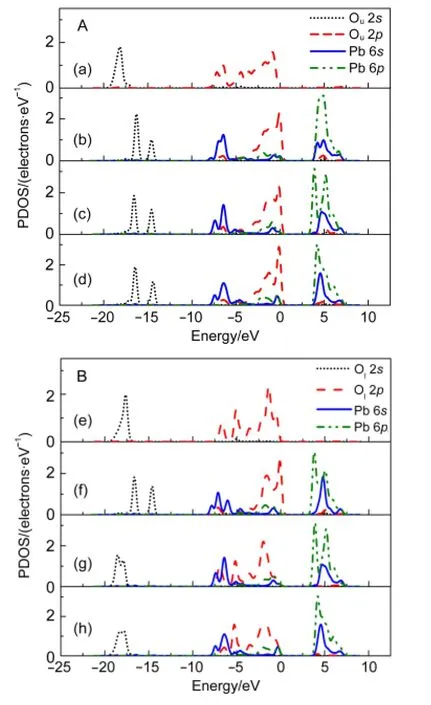
Fig.5 Partial electronic densities of states(PDOS)of Ouin the cleanAl(o)surface(a),Ouand Pb in configurationsA(b),C(c),D(d)(shown in partA)and Olin the cleanAl(o)surface(e),Oland Pb in configurations B(f),C(g),D(h)(shown in part B)
In the matter of Pb-Olinteraction(shown in Fig.5B),Ol2sin configuration B(f)was obviously different from those in configurations C(g)and D(h),but close to Ou2sin configuration A(b).Thus,Pb-Osinteractions in monodentate complexes were the same whatever at the“Ou”site or“Ol”site,different from those in bidentate complexes due to the competitive coordination.Ol2sand Ol2pin configurations C(g)and D(h)were similar to those of clean Al(o)surface(e),suggesting that Olcoordinated with hydrogen to a much larger extent than Pb(II).Affected by the dominant hydrogen coordination,overlaps of bonding orbitals between Pb 6sand Ol2pat-8.0--6.0 eV increased.Bond of Pb―Olexhibited small positive population value ultimately.
Additionally,no notable difference between two types of surface hydroxyls(OuH and OlH)has been found,as they have comparable Mulliken charges(-1.06 and-1.05)and similar DOS curves.Thus,the main reason for Pb(II)preferring to bind on the“Ou”site with relatively high CN and large binding energy is that it has the favorable position for hydrogen bonding interaction to stabilize the structure.As hydrogen bond of the“Ol…Hw”type plays a key role in determining stability of the complex,and distance between two nearest atoms of“Ol”is too large,Pb(II)is less likely to adsorb on the“Ol”site due to the strong repulsions of surrounding OuH to aqua ligands of Pb(II).
4 Conclusions
The structure and mechanism of Pb(OH)+adsorption on the Al(o)(001)kaolinite surface have been investigated by the plane-wave pseudopotential DFT calculations.Pb(OH)(H2O)5+is found as the most probable species of Pb(OH)+in aqueous system and used as the initial absorbate.All complexes in monodentate and bidentate modes feature the hemi-directed geometry with low CNs of 3 to 5,where the steric hindrance effect of kaolinite acts as the major cause.Hydrogen bonding interaction of surface“Ol”with“Hw”of aqua ligands has been found playing a key role in determining the stability of complex.Pb(II)adsorption in monodentate way prefers the“Ou”to“Ol”site as it has favorable position for the“Ol…Hw”interaction.Among the adsorption types examined,monodentate complex of“Ou”site with the highest binding energy may be the major species,and bidentate complex on“OuOl”site of single Al center is also probable.Upon the Mulliken population and DOS analysis,Pb(II)accepts electrons from surface oxygens during the adsorption process.Pb 6pcoupling with the Pb 6s―O 2pantibonding states is the primary orbital interaction of Pb(II)with surface oxygen.Pb-Olinteraction in the joint coordination structure features the predominantly bonding state fill-ing due to the competitive coordination of hydrogen.
Supporting Information:Atomic coordinates in initiative cell of kaolinite,equilibrium geometries of Pb(OH)(H2O)+1-5and[S(OH)2(O)Pb(OH)(H2O)m]with corresponding structural parameters have been included.The information is available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
(1) Karlsson,K.;Viklander,M.;Scholes,L.;Revitt,M.J.Hazard.Mater.2010,178,612.doi:10.1016/j.jhazmat.2010.01.129
(2)WasimAktar,M.;Paramasivam,M.;Ganguly,M.;Purkait,S.;Sengupta,D.Environ.Monit.Assess.2010,160,207.doi:10.1007/s10661-008-0688-5
(3)ATSDR(Agency for Toxic Substances and Disease Registry).Toxicological Profile for Lead(Update).U.S.Department of Health and Human Services,Atlanta,Georgia.http://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=96&tid=22(accessed Nov 20,2012).
(4) Tarasevich,Y.I.;Klimova,G.M.Appl.Clay Sci.2001,19,95.doi:10.1016/S0169-1317(01)00061-8
(5) Gupta,S.S.;Bhattacharyya,K.G.Phys.Chem.Chem.Phys.2012,14,6698.doi:10.1039/c2cp40093f
(6)Hong,H.L.;Min,X.M.;Zhou,Y.J.Wuhan Univ.Technol.2007,22,661.doi:10.1007/s11595-006-4661-2
(7) Spark,K.M.;Wells,J.D.;Johnson,B.B.Eur.J.Soil Sci.1995,46,633.doi:10.1111/ejs.1995.46.issue-4
(8) Srivastava,P.;Singh,B.;Angove,M.J.Colloid Interface Sci.2005,290,28.doi:10.1016/j.jcis.2005.04.036
(9) Hizal,J.;Apak,R.;Hoell,W.H.Environ.Prog.Sustain.2009,28,493.doi:10.1002/ep.v28:4
(10) Pearson,R.G.J.Am.Chem.Soc.1963,85,3533.doi:10.1021/ja00905a001
(11) Puskar,L.;Barran,P.E.;Duncombe,B.J.;Chapman,D.;Stace,A.J.Phys.Chem.A2005,109,273.doi:10.1021/jp047637f
(12) Shimoni-Livny,L.;Glusker,J.P.;Bock,C.W.Inorg.Chem.1998,37,1853.doi:10.1021/ic970909r
(13)Hummer,K.;Grüneis,A.;Kresse,G.Phys.Rev.B2007,75,195211.doi:10.1103/PhysRevB.75.195211
(14) Clark,S.J.;Segall,M.D.;Pickard,C.J.;Hasnip,P.J.;Probert,M.I.J.;Refson,K.;Payne,M.C.Z.Kristallographie2005,220,567.
(15) Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996,77,3865.doi:10.1103/PhysRevLett.77.3865
(16) Ireta,J.;Neugebauer,J.;Scheffler,M.J.Phys.Chem.A2004,108,5692.doi:10.1021/jp0377073
(17) Sun,T.;Wang,Y.B.Acta Phys.-Chim.Sin.2011,27(11),2553.[孙 涛,王一波.物理化学学报,2011,27(11),2553.]doi:10.3866/PKU.WHXB20111017
(18) Vanderbilt,D.Phys.Rev.B1990,41,7892.doi:10.1103/PhysRevB.41.7892
(19) Monkhorst,H.J.;Pack,J.D.Phys.Rev.B1976,13,5188.doi:10.1103/PhysRevB.13.5188
(20) Bish,D.L.Clay.Clay Miner.1993,41,738.doi:10.1346/CCMN
(21) Hu,X.L.;Michaelides,A.Surf.Sci.2008,602,960.doi:10.1016/j.susc.2007.12.032
(22) Kremleva,A.;Krüger,S.;Ro¨sch,N.Langmuir2008,24,9515.doi:10.1021/la801278j
(23) Mason,S.E.;Iceman,C.R.;Tanwar,K.S.;Trainor,T.P.;Chaka,A.M.J.Phys.Chem.C2009,113,2159.doi:10.1021/jp807321e
(24) Gourlaouen,C.;Gerard,H.;Parisel,O.Chem.-Eur.J.2006,12,5024.
(25)Wang,J.;Xia,S.W.;Yu,L.M.Acta Chim.Sin.2013,71,1307.[王 娟,夏树伟,于良民.化学学报,2013,71,1307.]
(26)Mishra,B.;Haack,E.A.;Maurice,P.A.;Bunker,B.A.Chem.Geol.2010,275,199.doi:10.1016/j.chemgeo.2010.05.009
(27) Bargar,J.R.;Brown,G.E.,Jr.;Parks,G.A.Geochim.Cosmochim.Acta1997,61,2617.doi:10.1016/S0016-7037(97)00124-5
(28) Bargar,J.R.;Brown,G.E.,Jr.;Parks,G.A.Geochim.Cosmochim.Acta1997,61,2639.doi:10.1016/S0016-7037(97)00125-7
(29)Walsh,A.;Watson,G.W.J.Solid State Chem.2005,178,1422.doi:10.1016/j.jssc.2005.01.030
(30) Mudring,A.V.Eur.J.Inorg.Chem.2007,2007(6),882.
猜你喜欢
高等理科教育(2022年2期)2022-05-09
化工职业技术教育(2021年5期)2021-11-09
化工职业技术教育(2021年1期)2021-03-15
科技进步与对策(2021年3期)2021-03-11
化工职业技术教育(2020年4期)2020-01-19
意林·全彩Color(2019年7期)2019-08-13
校园英语·中旬(2018年4期)2018-06-12
韩国语教学与研究(2017年2期)2017-03-07
化工学报(2016年3期)2016-03-14
天然产物研究与开发(2012年4期)2012-12-22
