
Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的几何结构、光电性质及磁性
2014-06-09 12:33:45智丽丽李艳青杨莲红赵高峰
计算物理 2014年6期
智丽丽, 李艳青, 杨莲红, 赵高峰
(1.伊犁师范学院新疆凝聚态相变与微结构实验室,伊宁 835000;2.新疆昌吉学院物理系,昌吉 831100;3.山东大学晶体材料国家重点实验室,济南 250100;4.河南大学物理与电子学院,开封 475004)
Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的几何结构、光电性质及磁性
智丽丽1,2, 李艳青2,3, 杨莲红2, 赵高峰4
(1.伊犁师范学院新疆凝聚态相变与微结构实验室,伊宁 835000;2.新疆昌吉学院物理系,昌吉 831100;3.山东大学晶体材料国家重点实验室,济南 250100;4.河南大学物理与电子学院,开封 475004)
基于密度泛函理论中的广义梯度近似系统研究Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的几何结构、光电性质和磁学性质.结果表明:Fe,Co原子相对于Ni原子更易于在(SiO2)3团簇上聚集;通过分析团簇的分裂途径及其产物,发现稳定性较好的氧化硅是一种很好的用于负载过渡金属“岛膜”的载体材料;Mn(SiO2)3团簇的能隙恰好位于近红外光谱范围内.通过磁性分析发现,该复合团簇的磁矩主要局域在过渡金属原子周围,而且,Fe2(SiO2)3和Co3(SiO2)3具有相对较大的磁矩,这主要源于过渡金属原子的d轨道间相互耦合.能隙和磁性两方面性质进一步肯定了二氧化硅磁性复合材料在医学界被用作光动力靶向治疗的可观前景.
Mn(SiO2)3团簇;几何结构;光电性质;磁性
0 引言
随着纳米技术的发展,人们发现纳米量级的物质具有与块体完全不同的性质,特别是作为团簇分子其物理化学性质随团簇的尺寸会发生明显的变化.硅的氧化物是地球上含量和用途较广的物质之一,尤其是二氧化硅在微电子、光通讯、和薄膜技术等许多领域中有着广泛的用途,因此,二氧化硅团簇和纳米颗粒方面的研究在近几年成为热点[1-3].有关报道指明(SiO2)n(n≤6)团簇在其小尺寸时,其基态结构是链状的构型[4-6].
目前,金属-氧化硅聚合物也是科学界关注的焦点,原因在于这些聚合物有着独特的性能,如:孙强等人[7]研究了Au原子与二氧化硅团簇的相互作用,他们指出该团簇体系可吸收近红外光,并将光转化为热量从而来杀死癌细胞.本课题组成员分别计算了Cu和Ag原子在二氧化硅团簇上的吸附,结果表明贵金属吸附二氧化硅均可以吸收近红外光,从而被用于癌细胞的治疗中[6,8].最近研究表明,(Fe\Co\Ni)/SiO2复合纳米微球不仅具有高的化学稳定性,大的比表面积,超顺磁性,还具有良好的生物相容性[7].这些性能使其具有极广阔的应用前景,因而在核酸提取,药物靶向等生物医学领域有着广泛的应用.值得关注的是:李雅丽[9]等人制备的二氧化硅磁性复合颗粒类似于贵金属二氧化硅复合材料,也可以吸收可见-近红外光,而且该微粒还具有超顺磁性,因此,该复合颗粒有可能被用于光动力靶向治疗中.据有关报道指出Ni/SiO2纳米复合材料也有着独特的光学和磁学性能[10].
鉴于(Fe\Co\Ni)/SiO2复合材料独特的光学和磁学特性,本文采用基于密度泛函理论中的广义梯度近似,从分子水平上系统地研究了Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的几何结构、光电性质及磁学性质,这将有利于从微观上理解这一复合体系所具有的光电磁特性,以及过渡金属与二氧化硅之间的相互作用.本文以(SiO2)3作为基体,原因在于实验上已经成功合成了(SiO2)3团簇分子[11],为设计新型纳米M/SiO2复合材料提供有力的的理论依据.
1 计算方法
计算采用密度泛函方法,选用广义梯度近似(GGA)泛函,利用Dmol3计算程序包完成[12].在电子结构计算中,采取全电子和包括d极化的双数基组,交换关联相互作用采用GGA-PW91方法自洽场,收敛标准为2.625 5×10-2kJ·mol-1.为了加速自洽场收敛我们使用了DIIS方法,轨道计算中使用的Smearing标准为13.125 kJ·mol-1.在几何优化过程中,力的收敛标准是52.5 kJ·mol-1·nm-1,位移收敛标准5.0×10-4nm,能量收敛标准为2.625 5×10-2kJ·mol-1,整个计算过程选取fine精确的计算标准.
为了验证方法的可靠性,在相同条件下,首先计算了O2、Si2、Fe2、Co2、Ni2二聚体以及Si-O、Fe-Si、Co-Si、Ni-Si的键长,并与已有的实验值或理论报道相对照,如表1所示,计算结果与已有报道吻合较好,表明我们所选计算方法是合理的.

表1 O2、Si2、Fe2、Co2、Ni2二聚体及Si-O、Fe-Si、Co-Si、Ni-Si的键长(单位:Å)Table 1 Bond length(in Å)of O2,Si2,Fe2,Co2,Ni2and Si-O,Fe-Si,Co-Si,Ni-Si
2 结果与讨论
2.1 几何结构
2.1.1 M(SiO2)3(M=Fe,Co,Ni)
图1给出了(SiO2)3和M(SiO2)3(M=Fe,Co,Ni)团簇的基态结构,其中,所有几何结构图中还标出了相应键长和原子的Mulliken电荷分布(括号中数值).如图1所示,M(SiO2)3(M=Fe,Co,Ni)团簇有着非常相似的基态结构,单个过渡金属原子均吸附在(SiO2)3末端的Si原子上,这是由于中间的Si原子是满配位的,而末端的两个Si原子配位数未满,因此,末端Si原子上的悬挂键为过渡金属Fe,Co,Ni原子的吸附提供了合适的位置.有趣的是单个过渡金属原子在(SiO2)3团簇上吸附的方式和单个贵金属原子在(SiO2)3团簇上吸附方式一致[6-8].
从图1可以看出,Fe-Si,Co-Si,Ni-Si的键长分别是2.177 Å,2.144 Å,2.130 Å,Fe-O,Co-O,Ni-O的键长分别是1.818 Å,1.826 Å,1.809 Å,M(SiO2)3(M=Fe,Co,Ni)团簇中与过渡金属原子相邻的末端Si-O键长分别是1.661 Å,1.649 Å,1.630 Å,较单纯(SiO2)3团簇末端Si-O键长(1.520 Å)均有所增加.在自由团簇中,由于处在不等价空间位置的原子感受到不同的势场,一部分原子将失去电荷,另一部分原子将得到电荷,从而出现电荷转移现象.Fe,Co,Ni分别失去了0.378 e,0.250 e,0.261 e,这些电荷主要引起与其临近的Si,O原子电荷的重新分布.
2.1.2 M2(SiO2)3(M=Fe,Co,Ni)
当第二个过渡金属原子被置于M(SiO2)3团簇的不同位置,经几何优化后发现,能量最低态中该原子吸附在原有的金属原子上,而且,三种团簇的基态结构非常相似(如图1所示).其中,Fe-Fe,Co-Co,Ni-Ni键长分别是2.192 Å,2.226 Å,2.239 Å,Fe-Si,Co-Si,Ni-Si的平均键长为2.391 Å,2.362 Å,2.344 Å.Fe2(SiO2)3中第二个Fe原子失去0.024 e,相对于Fe(SiO2)3团簇Fe2(SiO2)3中第一个Fe原子失去的电荷数几乎没变.Co2(SiO2)3中第二个Co原子得到0.033 e,Ni2(SiO2)3中第二个Ni原子得到较少的电子(0.017 e),在Ni2(SiO2)3中参与作用的主要仍是第一个Ni原子,该原子失去更多的电子,表明Fe,Co原子相对于Ni原子更易于在(SiO2)3团簇上聚集.
2.1.3 M3(SiO2)3(M=Fe,Co,Ni)
如图2所示,Fe3(SiO2)3和Co3(SiO2)3团簇的基态构型相似,第三个过渡金属原子与前两个过渡金属原子相结合,三个过渡金属原子形成了一个三角形吸附在(SiO2)3团簇上.不同的是Ni3(SiO2)3中的第三个Ni原子吸附在另一端的Si原子上,进一步表明二氧化硅团簇上Fe,Co原子比Ni原子更易于形成“岛膜”,这是由于Ni原子的外层电子排布为3s23p63d10的闭壳层排布,不利于与更多金属原子参加团聚作用,而Fe,Co原子的外层电子排布均为开壳层排布,易于发生电子间的相互转移,从而促进了更多外来原子的聚集.
2.1.3 混作和套种 混作和套种是指在同一个有机农作物种植环境中多样化种植有机农作物,以此防治害虫对其侵害。例如,在棉花田里种植高粱等能控制棉铃虫产卵的农作物,以实现集中诱杀害虫的目的,保护棉花作物不受棉铃虫的侵害。
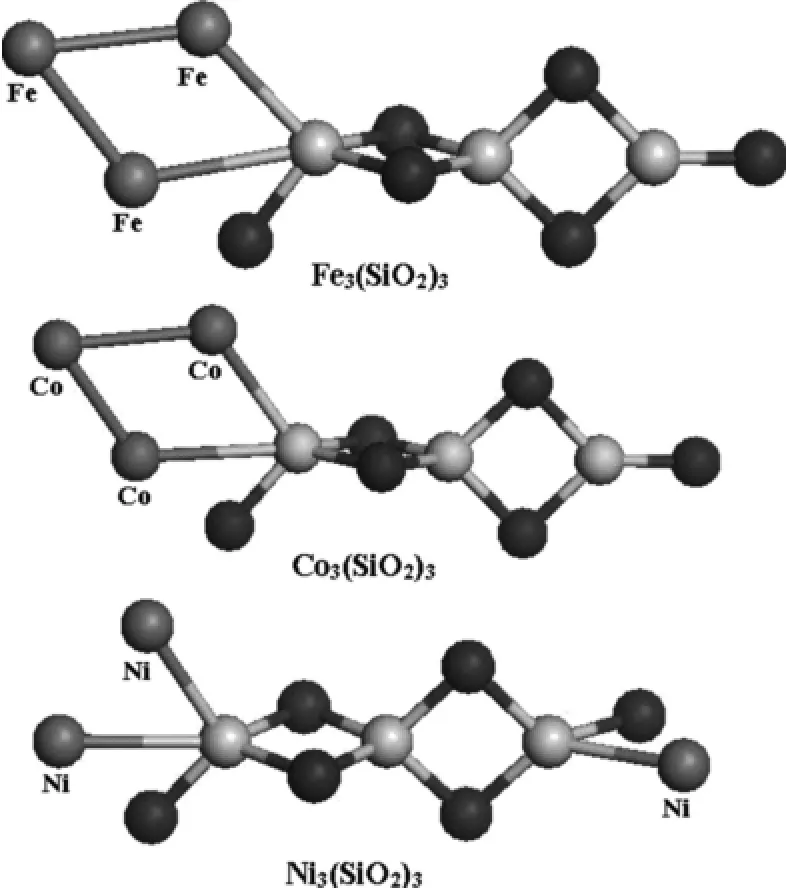
图2 M3(SiO2)3(M=Fe,Co,Ni)团簇的基态结构(黑色球为O,灰色球为Si)Fig.2 Ground state structures of M3(SiO2)3(M=Fe,Co,Ni)clusters
为了研究团簇的稳定性,我们给出Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的平均结合能,平均结合能的定义是团簇的结合能除以团簇的总原子数,其表达式:
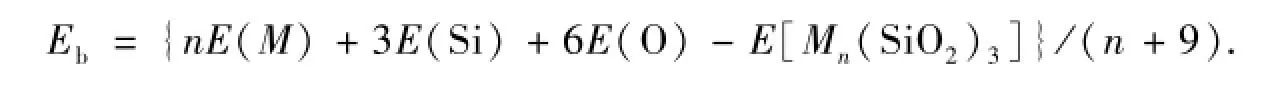
由表2可以看出,三种不同过渡金属元素和不同原子数目的原子吸附在氧化硅团簇上构成的复合体几乎具有相近的平均结合能(4.810 eV~5.354 eV),这与它们具有非常相似的几何结构有关,表明氧化硅是一种很好的用于负载过渡金属“岛膜”的载体材料.

表2 Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的优选分裂途径、分裂能(Ed)、平均结合能(Eb)、能隙、团簇总磁矩和金属原子磁矩(Mn)Table 2 Optimal disaggregated way,disaggregated energy,average binding energy,energy gap and moment of Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)clusters
通过分析团簇的分裂途径及其产物,便于我们理解表面吸附或沉积等现象.表2给出了Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的优选分裂途径及其相应的分裂能,分裂能定义:如果分裂途径为AmBn→Am+Bn,则分裂能定义为Ed=E(Am)+E(Bn)-E(AmBn),其中,E(Am)、E(Bn)和E(AmBn)均代表相应团簇的基态能量.Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的优选分裂产物中均包含有(SiO2)3团簇,另一种产物是过渡金属原子的聚集体,这表明链状的(SiO2)3团簇具有很好的稳定性,这与之前的文献报道相一致[4].而且,随着金属原子数目的增加,聚集体Mn内部的团聚力不断增加,这将有利于过渡金属“岛膜”的生长.
团簇一个很重要的电子性质就是其能隙,即电子最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)之间的能量差,如表2所示,除Con(SiO2)3(n=1-3)之外,Mn(SiO2)3(M=Fe,Ni;n=1-3)团簇随着金属原子数目的增加,能隙不断减小,发生红移现象.而且,从整体看,该复合团簇的能隙介于0.786 eV~1.131 eV,恰好位于近红外光谱范围内,与已有的贵金属吸附在(SiO2)n团簇上结果一致[6-8],说明Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇可以吸收近红外光,并将光转化为热量来杀死癌细胞,可能是一种治疗肿瘤和癌症的新型功能材料,同时,Fe,Co,Ni均是人体不可缺少的微量元素,我们的计算结果进一步肯定了过渡金属—二氧化硅磁性复合材料在医学界被用作光动力靶向治疗的可观前景.
为了进一步分析过渡金属在(SiO2)n团簇上聚集的机理,图3和图4分别给出了Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇分子的前线轨道分布,由图可知,电子云主要密集在过渡金属原子周围,这些原子将有利于与外来原子发生相互作用,因此,Fe,Co,Ni原子均在(SiO2)n团簇上发生了聚集现象,与前面的基态结构图保持一致.但是,Ni原子在二氧化硅上的吸附状态却不同于Fe和Co,由图4可知Ni2(SiO2)3的HOMO轨道发生凹陷,即Ni原子的d轨道发生了畸变,此时,第三个Ni原子主要受悬挂键的影响吸附到另一端的Si原子上.总体来看,过渡金属原子在(SiO2)n团簇上的吸附不仅受悬挂键的影响,还受过渡金属原子外层电子排布和分子前线轨道分布的影响.
为了进一步研究该复合体系的电子性质,计算了Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的分波态密度,图5是Con(SiO2)3(n=1-3)团簇的自旋分波态密度,从总体来看,在费米面附近起主要贡献的是p、d轨道,而且,随着过渡金属原子数目的增加,d轨道贡献不断增强,p轨道却显著下降,表明该分子轨道主要是由过渡金属原子的3d轨道提供的.
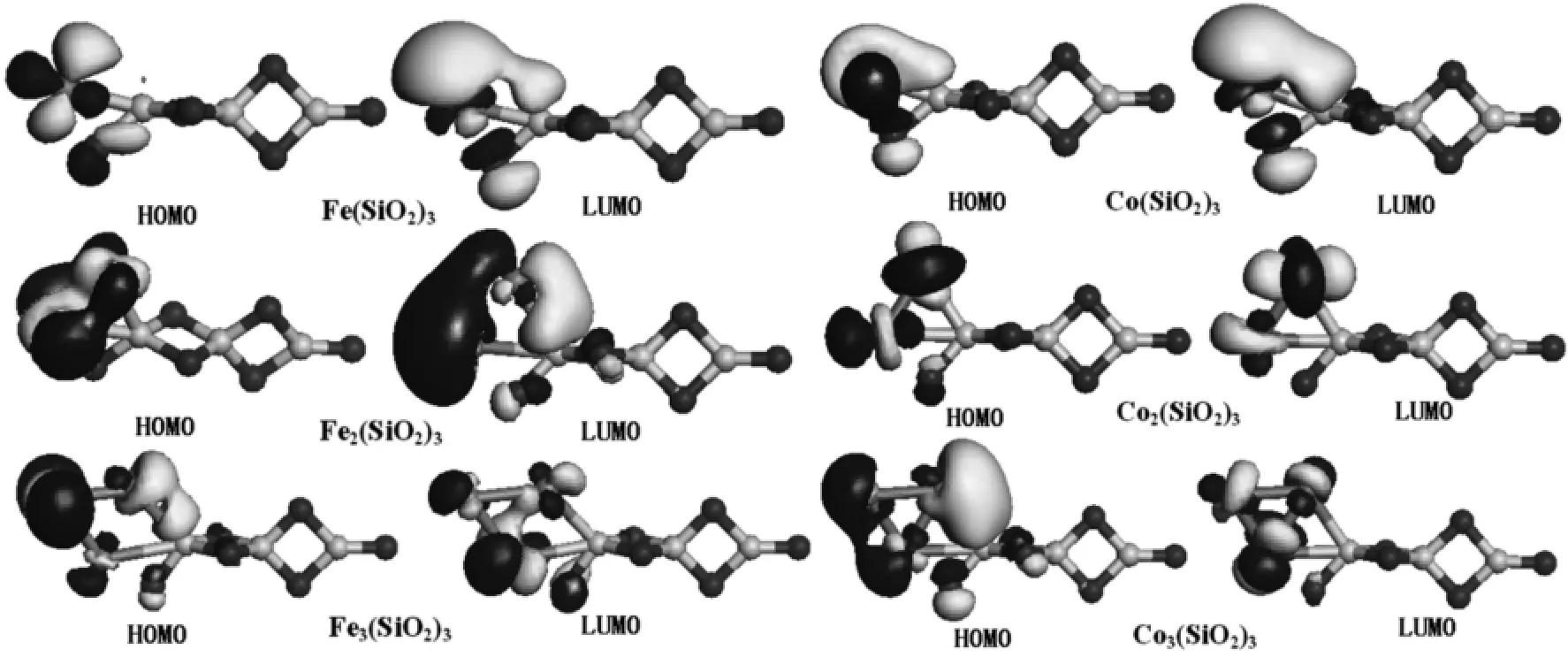
图3 Fen(SiO2)3和Con(SiO2)3(n=1-3)团簇的HOMO和LUMO轨道Fig.3 HOMO和LUMO orbitals of Fen(SiO2)3和Con(SiO2)3(n=1-3)clusters

图4 Nin(SiO2)3(n=1-3)团簇的HOMO和LUMO轨道Fig.4 HOMO和LUMO orbitals of Nin(SiO2)3(n=1-3)clusters
2.3 磁学性质
对于含有过渡金属的团簇,我们关心的另外一个问题是团簇的磁性.表2给出了基态Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的总磁矩和过渡金属原子的磁矩,原来不具有磁性的氧化硅团簇在吸附有过渡金属后变成了很好的磁性团簇,表明Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇内部出现了显著的自旋极化现象,而且,由表2可知团簇的磁矩主要是由过渡金属原子提供的.
Fe(SiO2)3中Fe原子磁矩近似于铁块体材料所具有得磁矩(2.22 μB[13]),Fe2(SiO2)3中两个Fe原子磁矩均大于铁块体材料的磁矩,使该团簇具有6 μB的磁矩,实验已经表明由铁磁性原子(Fe,Co,Ni)构成的团簇体系的磁矩远大于其块体材料的磁矩[14-15].Fe3(SiO2)3的总磁矩为4 μB,第三个Fe原子出现了反铁磁性(磁矩为-3.027 μB).Co2(SiO2)3和Co3(SiO2)3团簇也具有较高的磁矩分别是4 μB、7 μB,其中,该复合团簇中每个Co原子的磁矩均大于钴块体材料的磁矩(1.72 μB[13]).Fe2(SiO2)3和Co3(SiO2)3具有较大的总磁矩可能是由于过渡金属原子的d轨道之间发生显著耦合,加强了电子的自旋极化,与图3中展示的电子云分布情况保持一致.
值得注意的是Ni(SiO2)3团簇出现磁矩淬灭现象,主要原因是其中的Ni原子磁矩为零,而且,该团簇中其它原子的磁矩也均为零,Ni2(SiO2)3和Ni3(SiO2)3团簇的磁矩都是2 μB,吸附不同数目的Ni原子却导致相同的磁矩,主要是因为Ni3(SiO2)3中吸附在末端的第三个Ni原子发生了磁矩淬灭现象.
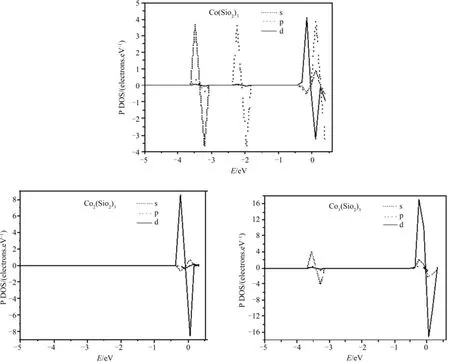
图5 Con(SiO2)3(n=1-3)团簇的分波态密度Fig.5 Partial density of states for Con(SiO2)3(n=1-3)clusters
为了进一步研究过渡金属-二氧化硅复合团簇的磁学性质,在考虑不同构型和不同自旋多重度的情况下,给出了Mn(SiO2)3(M=Fe,Co,Ni;n=2-3)团簇的铁磁(FM)和反铁磁(AFM)能量差,对于M2(SiO2)3(M=Fe,Co,Ni)团簇来说,分别是0.638 eV、0.490 eV、1.074 eV,M3(SiO2)3(M=Fe,Co,Ni)相应能量差为0.249 eV、0.265 eV、0.299 eV,这表明M2(SiO2)3(M=Fe,Co,Ni)相对于M3(SiO2)3(M=Fe,Co,Ni)团簇的铁磁性更加稳定.
3 结论
基于密度泛函理论中的广义梯度近似系统地研究了Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇的几何结构、电子性质和磁学性质.通过对基态结构、电子性质和磁学性质的分析,主要结论如下:
1)Fe,Co原子相对于Ni原子更易于在(SiO2)3团簇上聚集,而且,过渡金属原子在(SiO2)n团簇上的吸附不仅受悬挂键的影响,还受过渡金属原子外层电子排布和分子前线轨道分布的影响.
2)通过平均结合能、优选分裂途径及分裂能计算,从理论上证明了链状的(SiO2)3团簇具有很好的稳定性,是一种很好的用于负载过渡金属“岛膜”的载体材料.
3)Mn(SiO2)3(M=Fe,Ni;n=1-3)团簇的能隙介于0.786 eV~1.131 eV,恰好位于近红外光谱范围内,表明该复合材料可能是一种治疗肿瘤和癌症的新型医用功能材料.
4)Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)团簇内部出现了显著的自旋极化现象,原来不具有磁性的氧化硅团簇在吸附有过渡金属后变成了很好的磁性团簇,该磁矩主要局域在过渡金属原子周围,Fe2(SiO2)3和Co3(SiO2)3具有相对较大的磁矩(6 μB、7 μB).
[1] Wang L S,Nicholas J B,Dupuis M,et al.Si3Oy(y=1-6)clusters:Models for oxidation of silicon surfaces and defect sites in bulk oxide materials[J].Phys Rev Lett,1997,78:4450-4453.
[2] Li S,Silvers S J,El-Shall M S.Surface oxidation and luminescence properties of weblike agglomeration of silicon nanocrystals produced by a laser vaporization-controlled condensation technique[J].J Phys Chem B,1997,101(10):1794-1802.
[3] Harkless J A,Stilinger D K,Stillinger F H.Structures and energies of SiO2clusters[J].J Phys Chem,1996,100(4):1098-1103.
[4] Nayak S K,Rao B K,Khanna S N,et al.Atomic and electronic structure of neutral and charged SinOmclusters[J].J Chem Phys,1998,109(4):1245-1250.
[5] Zhi L L,Zhao G F,Guo L J,Jing Q.Structural,electronic,and vibrational properties of water molecules adsorbed on silica clusters[J].Phys Rev B,2008,77:235435/1-7.
[6] Zhao G F,Zhi L L,Guo L J,Zeng Z.The structural and electronic properties of Ag-adsorbed(SiO2)n(n=1-7)clusters[J].J Chem Phys,2007,127:234705/1-7.
[7] Sun Q,Wang Q,Rao B K,et al.Electronic structure and bonding of Au on a SiO2cluster:A nanobullet for tumors[J].Phys Rev Lett,2004,93(18):186803/1-4.
[8] 孙建敏,赵高峰,王献伟,等.Cu吸附(SiO2)n(n=1-8)团簇几何结构和电子性质的密度泛函研究[J].物理学报,2010,59(11):7830-7837.
[9] Li Y L,Yang M L.二氧化硅磁性复合微球的制备[D].西安:西北大学,2010.
[10] Yeshchenko O A,Dmitruk I M,Alexeenko A A,Dmytruk A M.Optical properties of sol-gel fabricated Ni/SiO2glass nanocomposites[J].J Phys Chem Solids,2008,69:1615-1622.
[11] Wang L S,Desai S R,Wu H,Nicholas J B.Small silicon oxide clusters:Chains and rings[J].Z Phys D,1997,40:36-39.
[12] Delley B.From molecules to solids with the DMol3approach[J].J Chem Phys,2000,113(18):7756-7764.
[13] 葛桂贤,罗有华,张建玮.FMBen(FM=Fe,Co,Ni;n=1-12)团簇结构和电子性质[J].物理化学学报,2008,24(10):1891-1896.
[14] Billas I M L,Chatelain A,de Heer Walt A.Magnetism from the atom to the bulk for iron,cobalt and nickel clusters[J]. Science,1994,265:1682-1684.
[15] Apsel S E,Emment J W,Deng J,Bloomfield L A.Surface-enhanced magnetism in nickel clusters[J].Phys Rev Lett,1996,76:1441-1444.
[16] Lide D R.CRC handbook of chemistry and physics[M].CRC,Boca Raton,FL,2000.
[17] Kant A,Strauss B.Dissociation energies of diatomic molecules of the transition elements.II:Titanium,chromium,manganese,and cobalt[J].J Chem Phys,1964,41(12):3806-3808.
[18] Hales D A,Su C X,Li L,Armenstout P B.Collision-induced dissociation of Co+n(n=2-18)with Xe:Bond energies of cationic and neutral cobalt clusters,dissociation pathways,and structures[J].J Chem Phys,1994,100:1049-1057.
[19] Purdum H,Montano P A,Shenoyand G K,Morrison T.Extended-x-ray-absorption-fine-structure study of small Fe molecules isolated in solid neon[J].Phys Rev B,1982,25:4412-4417.
[20] Wu Z J,Su Z M.Electronic structures and chemical bonding in transition metal monosilicides MSi(M=3 d,4 d,5 d elements)[J].J Chem Phys,2006,124:184306/1-15.
[21] Lindholm N F,Burgh D J,Rothschopf G K,Sickaf-oose S M,Morse M D.Optical spectroscopy of jet-cooled NiSi[J].J Chem Phys,2003,118:2190-2196.
Structural,Photoelectric and Magnetic Properties of Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)Clusters
ZHI Lili1,2,LI Yanqing2,3,YANG Lianhong2,ZHAO Gaofeng4
(1.Xinjiang Laboratory of Phase Transitions and Microstructures of Condensed Matters,Yili Normal University,Yining 835000,China;2.Department of Physics,Changji College,Xinjiang,Changji 831100,China;3.State Key Laboratory of Crystal Materials,Shandong University,Jinan 250100,China;4.School of Physics and Electronics,Henan University,Kaifeng 475004,China)
Equilibrium geometries,electronic and magnetic properties of Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)clusters are systematically studied employing density functional theory with a generalized gradient approximation.It shows that Fe and Co atoms are easier to congregate on(SiO2)3cluster than Ni atoms.It is found that stabler silica is an excellent matrix materials to carry islands of transition-metals.Energy gaps of Mn(SiO2)3(M=Fe,Co,Ni;n=1-3)clusters lie in near infrared radiation region.In analysis of magnetism,it is found that their magnetic moments are mainly located on transition-metal atoms.Fe2(SiO2)3and Co3(SiO2)3have greater magnetic moments,owing to coupling between d orbits of transition-metal atoms.Energy gap and magnetic property affirm a considerable foreground of magnetic-mulriple silica used for photodynamic target therapy in medical stage.
Mn(SiO2)3cluster;geometrical structure;photoelectric property;magnetism
date: 2013-12-03;Revised date: 2014-04-06
O561
A
2013-12-03;
2014-04-06
新疆维吾尔自治区高校科研计划资助(XJEDU2013S42);新疆凝聚态相变与微结构实验室开放课题基金(XJDX0912-2010-05和XJDX0912-2010-07)及昌吉学院院级课题(2013YJYB002)资助项目
智丽丽(1984-),女,研究生,讲师,主要从事团簇结构和电子性质研究,E-mail:zhilili2010@sina.com
1001-246X(2014)06-0727-08
猜你喜欢
数学物理学报(2022年3期)2022-05-25 13:33:22
数学物理学报(2022年1期)2022-03-16 06:15:04
数学物理学报(2021年5期)2021-11-19 07:01:16
数学物理学报(2021年3期)2021-07-19 06:02:18
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
深圳大学学报(理工版)(2015年6期)2015-11-26 12:33:48
河南科技(2014年23期)2014-02-27 14:18:52
