
分子表面随机采样分析用于苯乙基噻唑硫脲衍生物抗艾滋病药物的3D-QSAR研究
2014-06-06 01:00仝建波李梦龙
原子与分子物理学报 2014年3期
仝建波,赵 翔,钟 黎,常 佳,李梦龙
(1.陕西科技大学化学与化工学院,西安710021;2.教育部轻化工助剂化学与技术重点实验室,西安710021;3.四川大学化学学院,成都610064)
1 引 言
由人类免疫缺陷病毒(HIV)引起的艾滋病(即获得性免疫缺陷综合症,AIDS)自1981年被发现以来,在全球迅速蔓延,其中速度最快的地区是非洲和亚洲,中国也属于艾滋病高发地区.艾滋病的迅速蔓延引起了各国政府和科研机构的高度重视,纷纷投入大量的人力物力进行艾滋病的预防和治疗研究.自1987年第一个抗艾滋病药物齐多夫定(AZT)问世以来,已经有30余种不同类型的药物问世.
非核苷类逆转录酶抑制剂(NNRTIs)是一类与核苷结构、作用机理迥异的特异性抑制HIV-1 RT的化合物,该类抑制剂具有结构多样、高效低毒以及可与其他药物协同作用等特性,而且与核苷类逆转录酶相比,还具有许多独特的优点,比如它们的活性形式不需要细胞内的磷酸化,因而在静止和活化的细胞或不同的细胞系中都具有广泛的活性;它们亦可与细胞外的逆转录酶结合,减少游离病毒颗粒的逆转录,从而降低病毒的传染性[1-2].因而对NNRTIs的研究一直是发现新型抗艾滋病药物的有效途径和重要方向之一.
噻唑硫脲化合物N-(2-苯乙基)-N`-(2-噻唑基)-硫脲(PETT)及其衍生物[2]作为一种典型的HIV NNRTIs,如果能通过进一步结构优化提高活性,有望成为一类新的高效抗艾滋病药物.而定量结构活性关系(QSAR)研究是药物设计的一种重要方法,它对于设计和筛选生物活性显著的药物以及阐述药物的作用机理等具有指导作用[3-5].因而构建这些化合物分子结构与生物活性之间定量相关模型,对于研究、设计和开发出高效抗艾滋病药物具有重要意义.
QSAR为药物设计和筛选提供了相对简便的途径,并受到了广泛的重视.其中分子结构表征(MSC)是QSAR的一个关键环节,当前流行的MSC技术大致可以分为基于二维结构的2D结构描述子和基于三维空间构型的3D结构描述子两大类.由于2D描述子很难再现配基与受体的空间契合方式,因此3D结构描述子正成为QSAR研究的主流.采用本实验室新提出的一种3D分子结构描述方法—分子表面随机采样分析(RASMS),得到用于表征药物分子结构特征的三维矢量描述子,将其用于PETT类抗艾滋病药物的QSAR研究,取得了较好的结果.
2 原理和方法
2.1 RASMS中的几个概念
2.1.1 蛋白质受体原子探针
由于大多数药物直接作用靶标是具有一定生物活性功能的肽和蛋白质分子,因此RASMS法使用20种标准天然氨基酸中的各类原子作为探针.考虑到具有不同杂化状态的原子往往因所处基团和区域不同而使其对活性贡献表现出一定的差异,进而以此得到8个探针原子.为了反映这些探针的性质,本文分别给其赋予了平均电荷指数、范德瓦耳斯指数和平均疏水指数的概念.
平均电荷指数(Mean charge index,MCI):探针原子电性特征取其出现于氨基酸中的平均电荷数.具体计算如下:首先使用Chemoffice 8.0自带数据库生成20个天然氨基酸的初始分子立体结构;并利用分子模拟软件HyperChem 7.5进行分子力学构象优化;其结果进一步应用Gaussian 98W量子化学计算软件在密度泛函水平基于广义梯度近似法最终优化得到分子三维结构,并采用Mulliken布居分析法以单点形式计算出原子的净电荷数量[6-8];自编程序计算每种探针原子在氨基酸中出现的平均电荷数作为其MCI值.
范德瓦耳斯指数(Van der Waals index,VWI):通常文献给出的是孤立原子Van der Waals半径,但实际原子由于其所处分子的化学微环境和自身杂化状态不同该半径有所变化,因此本文对其进行了调整,即使用经校正后的原子Van der Waals半径作为探针原子的范德瓦耳斯指数[9-10],VWI=Ch×RVDW*.
平均疏水指数(Mean hydrophobic index,MHI):类似于MCI,MHI取每类探针的疏水性在天然氨基酸中出现的平均值.这里以文献[11]所定义的原子溶解参数(atomic solvation parameter,ASP)作为疏水性量度.
2.1.2 虚拟受体可及表面
本文提出了虚拟受体可及表面(pseudo-receptor accessible surface,PRAS)的概念.当作为药物作用靶点的生物分子(蛋白质、核酸、糖类等)中所包含的原子可以抵达的该药物分子表面称为分子虚拟受体可及表面(pseudo-receptor accessible surface of molecule,PRASM),如果以上述划分蛋白质8类探针原子中的氢作为受体探针,其定义如下:利用单个氢原子球体在药物分子Van der Waals表面滚动其球心所经历的曲面则称为虚拟受体氢原子可及表面(H-PRASM).同理可以计算其余7种虚拟受体探针原子可及表面(图1).依照上述PRASM的计算方法可以定义孤立原子的原子虚拟受体可及表面(pseudo-receptor accessible surface of atom,PRASA),显然该表面是一个球面(图2),其半径为该原子与探针半径之和.可以看到药物分子中每个原子的PRASA可能有一部分参与形成该分子的PRASM.
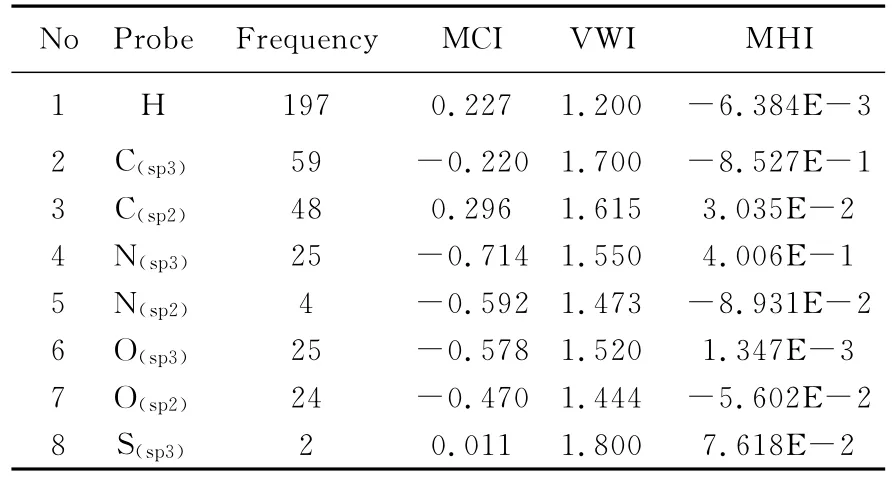
表1 8个探针原子在20种天然氨基酸中出现的频数及其MCI,VWI和MHI取值Table 1 The appearing frequencies of eight types of atomic probes in twenty types of natural amino acids as well as the values of its MCI,VWI and MHI
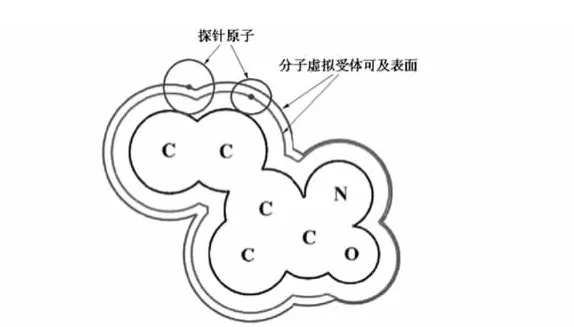
图1 分子虚拟受体可及表面Fig.1 Pseudo-receptor accessible surface of molecule
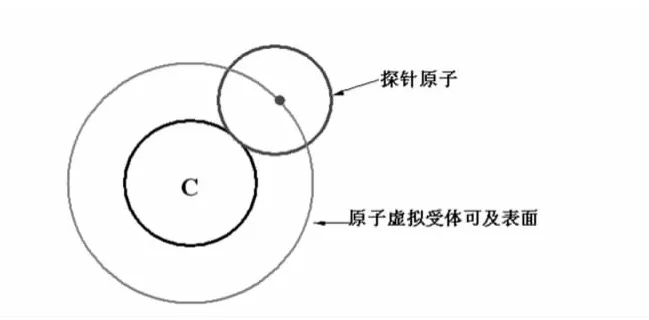
图2 原子虚拟受体可及表面Fig.2 Pseudo-receptor accessible surface of atom
2.2 药物分子中常见原子分类
有机化合物中常见的原子包括氢、碳、氮、磷、氧、硫、氟、氯、溴、碘,它们分别属于元素周期表的IA、IVA、VA、VIA、VIIA共计5个主族.基于“具有相同化学性质的原子应属于同一类”的观点,本文很自然的按照这些原子所处元素周期表的族将其划分为5大类.为了更好的表现分子细微结构特征,并考虑到同一族原子处于不同杂化状态时化学性质也有较大差异,继而在上述分类的基础上再进一步把不同族中的原子按其杂化状态细分为10类(表2).
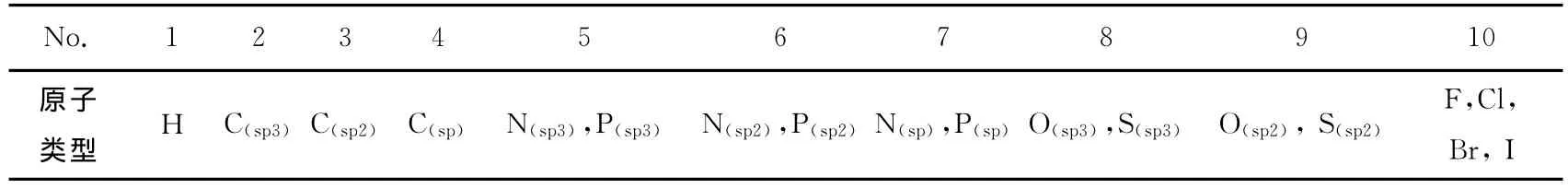
表2 RASMS中按周期表的族和原子杂化类型划分的10类原子Table 1 Ten types of atoms according to element periodic tables
2.3 探针原子与药物配体的作用模式
经典药学理论认为药物在抵达受体并与之发生作用绝大多数都是暂时的、可逆的非键效应,其表现为静电、立体、疏水、氢键、电荷转移等多种因素.本文考虑到静电、立体、疏水效果几乎包含了大部分这类信息,故在RASMS中主要计算这3种作用类型.对于氢键、电荷转移等可以看成是静电和立体效应的特殊表现形式.
静电作用(electrostatic interaction)是一类重要的非键效应,经典的点电荷作用方式服从Coulomb定理(式1).其中rij是探针到配体原子间的Euclid距离;e为单位电荷电量1.6021892×10-19C;ε0为真空中的介电常数8.85418782×10-12C2/J·m;Z为配体原子净电荷数;p和l分别为探针和配体原子所属类型.
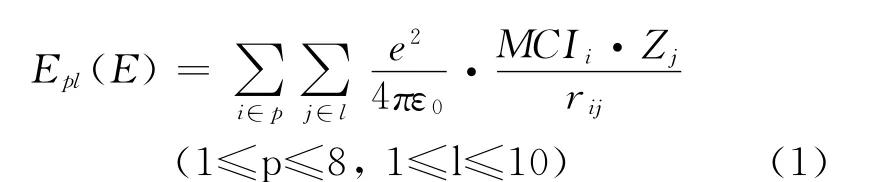
立体作用(steric interaction)是空间原子间存在的非偶极-偶极或偶极诱导作用,这里采用Lennard-Jones方程来描述这种作用方式(式2).该式中εij=(εii·εjj)1/2为探针-受体原子势能阱深,取文献值[12];D为经验推导的原子间作用能修正常数,取0.01;Rij*=(VWIi+Ch·Rj*)/2,为经过校正后的探针-受体原子间van der Waals碰撞半径,其校正因子Ch,当sp3杂化时取1.00,sp2杂化取0.95,sp杂化取0.90[13].
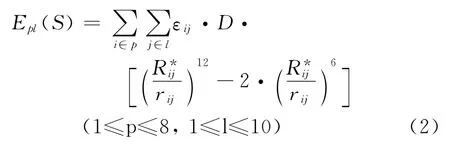
疏水作用(hydrophobic interaction)是影响药物分子与生物体结合的重要因素,由于其往往表现于体系熵的改变,因此很难用一个统一的公式来描述.对于有关疏水作用的研究已有众多文献报道,考虑到RASMS要求深入到配基分子内部原子与其表面受体探针相互作用,我们使用Kellogg等人提出的 HINT方法[14-18]来表达该类势能形式.在HINT中定义了一个简单的计算两个原子之间的疏水相互作用表达式(式3),该式中S为原子溶剂可及面积(solvent accessible surface area of atom,SASA),是水分子在原子表面滚动其球心形成的表面面积[19];a为原子疏水性常数,这里同样使用上文所提到的ASP作为其表达值;T是作用形式的二值判别函数,以表明不同类型原子疏水作用的熵效应变化方向.
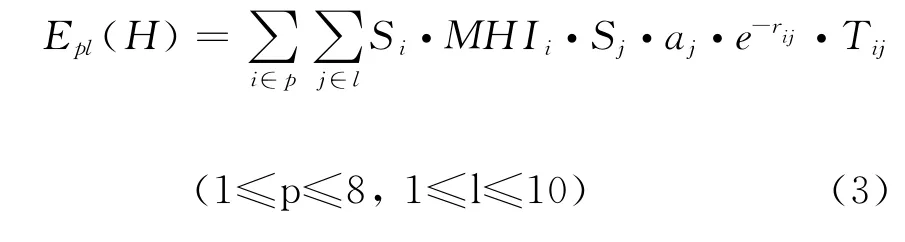
2.4 RASMS实现过程
使用Chemoffice 8.0分子图形构建软件包自动生成初始药物分子立体结构;同时应用MOPAC 6.0[20-21]半经验量子化学计算软件在 AM1(Austin model 1)[22]水平上采用共扼梯度法完成几何优化,并在最终分子结构上进行单点计算以求得每个原子所带电荷的Mulliken布居数;继而利用自编程序Sampling-tool进行RASMS分析:当以氢原子为探针时,首先在药物分子虚拟受体氢原子可及表面(H-PRASM)上进行随机取点作为该探针的探点,每一次采样都计算该点探针与药物分子中10类原子的相互作用情况,而每种作用又分为静电、立体和疏水3种效应,这样可以得到30个作用项,它包含了分子表面该点的势场分布状况.可以看到,经过大规模重复上述随机采样,探点将几乎完全均匀的覆盖分子周围,从而得到整个表面的场能分布信息.完成所有采样之后将多次探测得到的30项作用每项对应加和,并取其平均值作为该分子表面势场分布密度.其中第一项表示HPRASM上药物分子中第1类原子(氢原子)与探针平均作用情况,第二项表示第2类原子(sp3杂化的碳原子)与探针平均作用情况,以此类推.这样通过使用8个探针对一个药物分子表面进行采样计算将得到8×30=240个作用分量,即为RASMS法对该分子的最终分析结果(图3).

图3 RASMS法对于每一个药物分子得到的240个作用分量示意图Fig.3 240types of interaction with RASMS method of every drug molecule
3 结果与讨论
3.1 数据集的选取
61个PETT类衍生物的结构及活性数据来自文献[23](表3).活性标度为化合物对HIV-1感染细胞的半数有效抑制浓度IC50的负对数pIC50:
3.2 模型验证
为了深入研究RASMS方法与此类抗艾滋病药物活性的内在联系,采用多元线性回归(MLR)这种典型的线性方法进行建模.另外对所建模型的外部预测能力和真实有效性进行验证是定量构效关系中非常重要的一个部分,用来表征模型稳定性与预测能力的参数有rm、r0和k[24-27]等,检测方法在文献[24]中提到.为此,本文将数据集分成了51个内部样本和10个外部样本,10个外部样本将用于模型检验.
3.3 结构表征
使用Chemoffice 8.0自动搭建61个PETT类抗艾滋病药物分子的立体结构,用Chem3D自带的MOPAC半经验量子化学软件在AM1水平上最终优化得到分子结构(截断值0.0001kJ/mol),并采用Mulliken布居分析法以单点形式计算出原子的净电荷数量,将分子中每个原子的空间位置及电荷分别以笛卡尔坐标和净电子数目的形式输入实验室自编的应用程序Sampling-tool加以处理,得到分子的RASMS描述子.
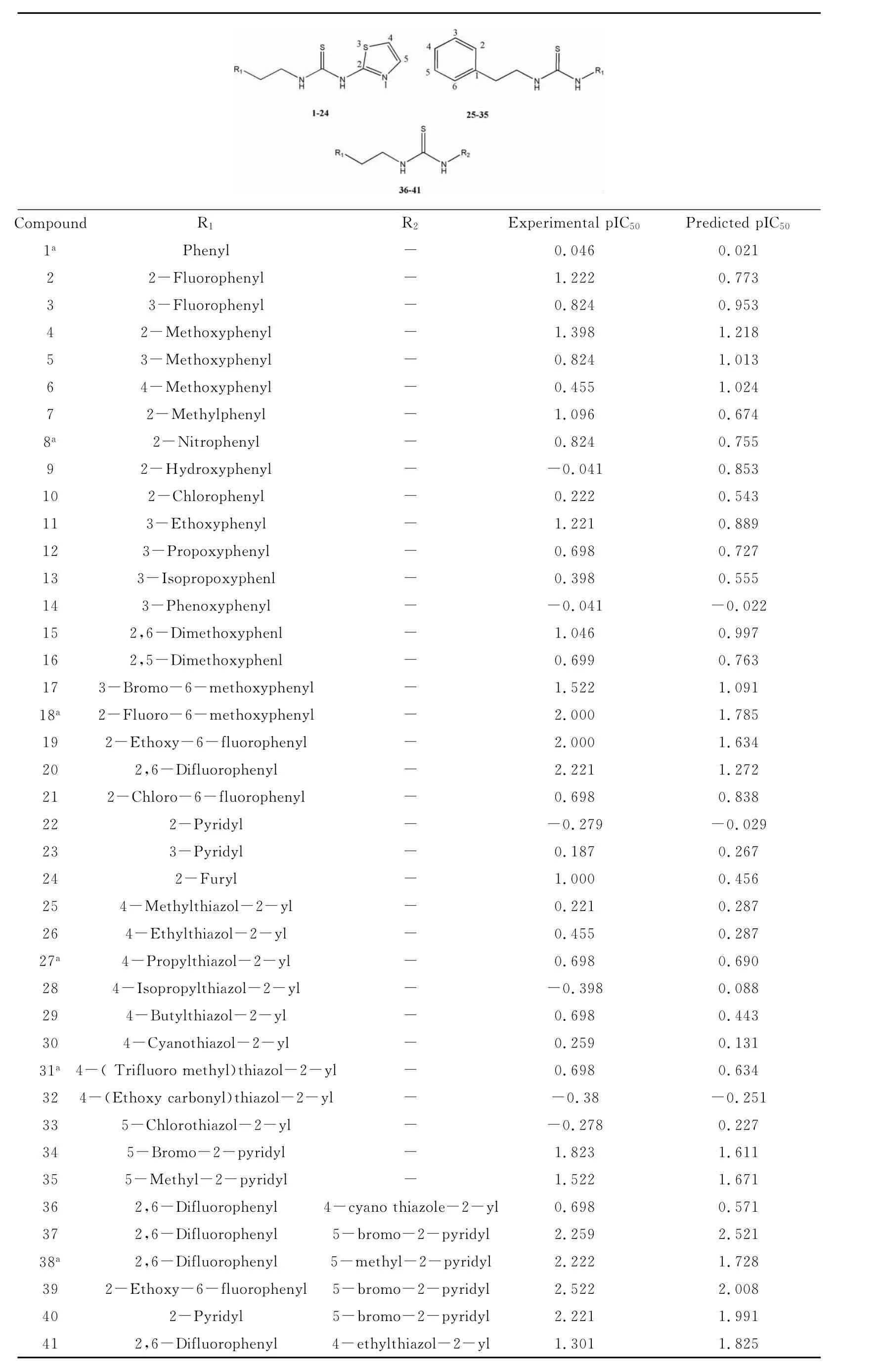
表3 苯乙基噻唑硫脲衍生物母体结构及其pIC50Table 3 Structure and pIC50activity of phenylethylthiazolylthiourea derivatives
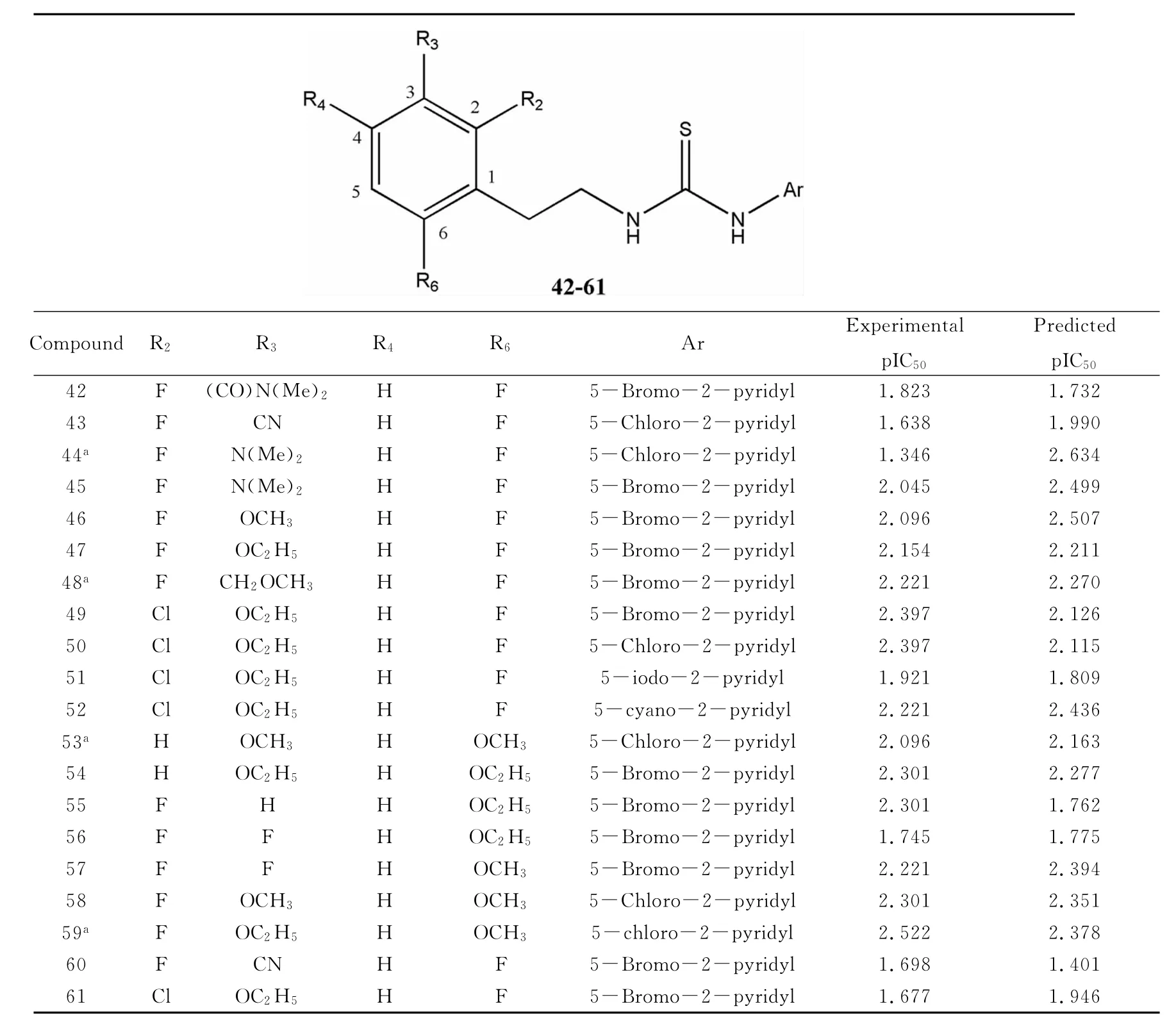
(continued)
3.4 多元线性回归建模
RASMS含有240个描述子,所研究51个PETT内部样本通过计算最终得到192个非零矢量,使用SPSS 18.0对这192个非零矢量进行逐步线性回归(stepwise multiple regression,SMR),并采取有进有出的原则,SMR按Fisher显著性检验依次引入变量,选出15个显著性矢量;同时将SMR每一步得到的原始变量多元线性回归(Multiple Linear Regression,MLR)建模,并使用留一法交互校验法计算模型的QCV大小,以其达到最大值时变量数目来确定最终模型.表征模型稳定性与预测能力的参数有rm、r0和k,定义如下:

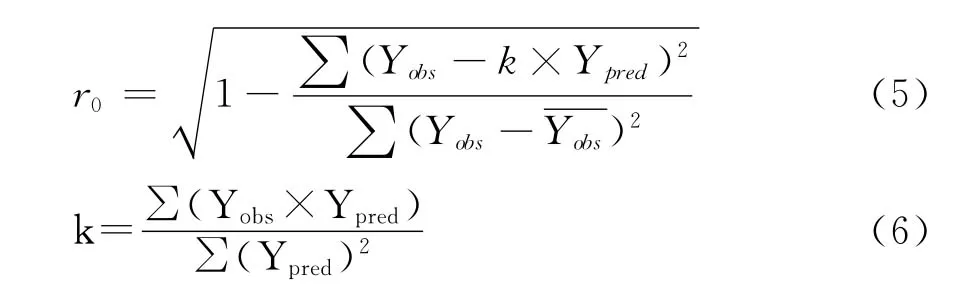
结合交互校验的复相关系数(QCV)变化情况,最终确定选用11变量回归模型,按照文献[22]的方法分别输入内部样本和外部样本前11个描述子和实验值即可得到:Rcum、QCV、r0(test)、rm(test)、rm(all)和k分别为0.919、0.856、0.936、0.848、0.837和0.967(0.85<k<1.15),模型的标准化形式如下:
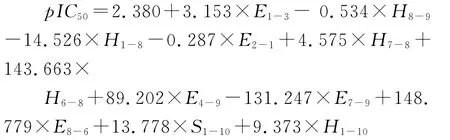
其中,E表示静电作用,S表示立体作用,H表示疏水作用,如,E1-3表示第1类探针与第3类原子之间的静电作用项,S1-10表示第1类探针与第10类原子之间的立体作用项,H8-9表示第8类探针与第9类原子之间的疏水作用项,以此类推.样本活性实验观测值与模型计算相关情况如图4,几乎所有样本都均匀分布于过原点45°直线周围,无明显异常点,结果表明RASMS能够较好地表征香豆素类化合物的活性与结构之间的关系.
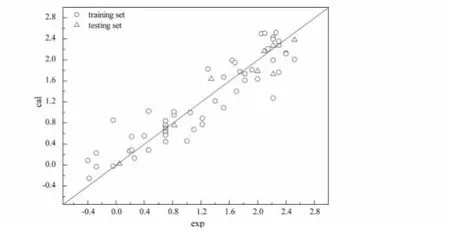
图4 苯乙基噻唑硫脲衍生物活性实验值与计算值关系图Fig.4 Plot of calculated versus observed pIC50for PETT derivatives with MLR
4 结 论
本文所建模型包含了静电、立体、疏水3种经典作用项,说明RASMS能够较好地表征有机化合物活性与环境间复杂相互作用,所建模型以化合物本身的立体参数与电性参数为基础.与当今大多3D药物设计技术相比,RASMS在应用过程中所建的模型物化意义明确,可解释性强,参数简单易得.本文使用RASMS对61个PETT类化合物进行了建模和定量构效关系研究,结果表明RASMS描述子与分子生物活性呈线性相关,具有一定的指导意义,可应用于潜在和新型抗艾滋病药物的设计和开发,值得进一步深入研究.
[1]Zhan P.Design,synthesis and anti-HIV evaluation of novel arylazolyl(azinyl)thioacetanilides as potent NNRTIs[D].China Shandong University,2010(in Chinese)[展鹏.新型芳唑(嗪)巯乙酰胺类HIV-1 NNRTI的设计、合成与活性研究 [D].中国:山东大学,2010]
[2]Tong J B,Wang P,Che T,etal.Studies of the quantitative structure activity relationship for phenylethylthiazolythiourea of anti-HIV drug using new three-dimensional structure descriptors[J].J.At.Mol.Phys.,2012,29(6):960(in Chinese)[仝建波,王平,车挺,等.苯乙基噻唑硫脲衍生物抗艾滋病药物3D-QSAR研究[J].原子与分子物理学报,2012,29(6):960]
[3]Zhang X,Yang L Y,Zheng Y T.Advances of screening methods in vitro for HIV-1intergrase in-hibitors[J].Chin.Pharmacol.Bull.,2013,29(1):14(in Chinese)[张璇,杨柳萌,郑永唐.HIV-1整合酶抑制剂体外筛选方法研究进展[J].中国药理学通报,2013,29(1):14]
[4]Xu Y S,Zhong R G,Zeng C C,etal.The Research progress of diketo Acid HIV Integrase Inhibitors[J].J.BeijingUniv.Technol.,2005,31(6):635(in Chinese)[徐义生,钟儒刚,曾程初,等.二酮酸类HIV整合酶抑制剂研究进展[J].北京工业大学学报,2005,31(6):635]
[5]Zhang N,Zhang H B,Huang S.The research progress of anti-HIV integrase inhibitors[J].Pharm.Biotechnol.,2009,16(3):269(in Chinese)[张楠,张惠斌,黄山.抗HIV整合酶抑制剂研究进展[J].药物生物技术,2009,16(3):269]
[6]Hohenberg P,Kohn W.Inhomogeneous electron gas[J].Phys.Rev.,1964,136:864.
[7]Kohn W,Sham L J.Self-consistent equations including exchange and correlation effects [J].Phys.Rev.,1965,140:1133.
[8]Mulliken R S.Electronic population analysis on LCAO-MO molecular wave functions.I [J].J.Chem.Phys.,1955,23:1833.
[9]Hahn M.Receptor surface models.1.definition and construction [J].J.Med.Chem.,1995,38:2080.
[10]Bondi A.Van der Waals volumes and radii[J].J.Chem.Phys.,1964,68:441.
[11]Pei J,Wang Q,Zhou J,etal.Estimating proteinligand binding free energy:Atomic solvation parameters for partition coefficient and solvation free energy calculation[J].Proteins:Structure,Functionand Genetics,2004,57:651.
[12]Hasel W,Hendrikson T F,Still W C.A rapid approximation to the solvent accessible surface areas of atoms[J].TetrahedronComp.Method,1988,1:103.
[13]Zhou P,Tong J B,Tian F F,etal.A novel comparative molecule/pseudo receptor interaction analysis[J].Chin.Sci.Bull.,2006,51(15):1824.
[14]Kellogg G E,Semus S F,Abraham D J.HINT-a new method of empirical hydrophobic field calculation for CoMFA [J].J.Comput.AidedMol.Des.,1991,5:545.
[15]Wireko F C,Kellogg G E,Abraham D J.Allosteric modifiers of hemoglobin.2.crystallographically determined binding sites and hydrophobic binding/interaction analysis of novel hemoglobin oxygen effectors[J].J.Med.Chem.,1991,34:758.
[16]Kellogg G E,Joshi G S,Abraham D J.New tools for modeling and understanding hydrophobicity and hydrophobic interactions[J].Med.Chem.Res.,1992,1:444.
[17]Kellogg G E,Abraham D J.KEY,LOCK,and LOCKSMITH.Complementary hydrophobicity map predictions of drug structure from a known receptor/receptor structure from known drugs[J].J.Mol.Graphics,1992,10:212.
[18]Nayak V R,Kellogg G E.Cyclodextrin-barbiturate inclusion complexes:a CoMFA/HINT 3-D QSAR study[J].Med.Chem.Res.,1994,3:491.
[19]Hasel W,Hendrikson T F,Still W C.A rapid approximation to the solvent accessible surface areas of atoms[J].TetrahedronComp.Method,1988,1:103.
[20]Stewart J J P.MOPAC:A semiempirical molecular orbital program [J].J.Comput.AidedMol.Des.,1990,4(1):1.
[21]Tong J B,Wang P,Che T,etal.Quantitative structure activity relationship studies of benzoxazinone of anti-HIV drug using new three-dimensional structure descriptors.[J].J.At.Mol.Phys.,2012,29(5):777(in Chinese)[仝建波,王平,车挺,等.苯并噁嗪酮衍生物类抗艾滋病药物3D-QSAR研究[J].原子与分子物理学报,2012,29(5):777]
[22]Dewar M J S,Zoebisch E G,Healy E F,etal.AM1:A new general purpose quantum mechanical molecular model[J].J.Am.Chem.Soc.,1985,107:3902.
[23]Razieh S,Afshin F,Behzad M.QSAR study of PETT derivatives as potent HIV-1reverse transcriptase inhibitors[J].J.Mol.GraphicsModell.,2009,28:146.
[24]Roy K,Chakraborty P,Mitra I,etal.Some case studies on application of“r2m♂”metrics for quality of quantitative structure-activity relationship predictions:emphasis on scaling of response data[J].J.Comput.Chem.,2013,34:1071.
[25]Mitra I,Saha A,Roy K.Exploring quantitative structure-activity relationship studies of antioxidant phenolic compounds obtained from traditional Chinese medicinal plants[J].Mol.Simul.,2010,13:1067.
[26]Roy K,Mitra I,Kar S,etal.Comparative studies on some metrics for external validation of QSPR models[J].J.Chem.Inf.Model.,2012,52:396.
[27]Roy K,Mitra I.On various metrics used for validation of predictive QSAR models with applications in virtual screening and focused library design [J].Comb.Chem.HighThroughputScreening,2011,14:450.
猜你喜欢
无机化学学报(2022年9期)2022-09-16
青春期健康(2021年23期)2021-12-14
装备制造技术(2020年1期)2020-12-25
中国生殖健康(2019年12期)2019-01-07
中国生殖健康(2019年5期)2019-01-06
现代检验医学杂志(2016年3期)2016-11-15
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
中国塑料(2016年11期)2016-04-16
现代检验医学杂志(2014年5期)2014-02-02
