
细胞色素P450蛋白酶催化下环己胺脱氨基反应机理的理论研究
2014-06-06 01:00:28张笑乾李朝正刘玉芳
原子与分子物理学报 2014年3期
张笑乾,李朝正,刘玉芳
(河南师范大学物理与电子信息工程学院,新乡453007)
1 引 言
细胞色素P450蛋白酶(简称P450)是一类血红素蛋白的超家族,被誉为“世界上最万能的生物催化剂”,因其在药物等外源化合物的代谢以及性激素等内源化合物的生物合成过程中的重要催化作用,而被越来越多的人所熟知.这一类单加氧酶可以参与催化众多化学反应,诸如非活泼碳氢化合物的羟基化反应、脱烷基反应以及脱氨基反应等等[1].其中,脱氨基反应中的机理细节,尤其是涉及碳氢键氧化过程的一些机理细节在过去几十年中引起了相当大的争议,虽然大量基于此过程的实验数据和研究结果已被报道,但仍未能完全解决这个颇具争议性的问题[2-6].因此,针对相关机理的理论计算,不仅可以揭示反应机理细节,对相关实验结果给出合理解释,还可以使发现新的机理特征成为可能,从而进一步指导实验进行更深入的研究.本文以细胞色素P450蛋白酶催化下环己胺的脱氨基反应为例,对其反应机理细节进行了研究.
图1所示为细胞色素P450催化环己胺脱氨基反应的整个反应过程,其中关于Cα-H键的氧化过程涉及两个假设机理.反应的第一步是环己胺转化为相应的碳自由基中间体(Ⅰ-Ⅱ或Ⅲ过程),随后自由基中间体经过一个氧反弹过程[7-8](Ⅳ过程)生成1-羟基-环己胺,接下来1-羟基-环己胺进一步分解(Ⅴ过程)成为环己酮和氨气,整个分解过程可以通过一个水分子的辅助,在非酶环境中进行.关于机理细节的争议主要集中在环己胺转变为碳自由基中间体这一步.其中一个机理是单电子传递(SET)机理[6](Ⅰ-Ⅱ过程),首先一个电子从环己胺的氮原子上传递到酶活性中心(Ⅰ过程),生成氨基阳离子自由基中间体,然后这个中间体再向酶活性中心传递一个质子(Ⅱ过程),至此生成一个碳自由基中间体;与之不同的另外一个机理是氢原子传递(HAT)机理[2-5](Ⅲ过程),即一个氢原子由环己胺传递到酶活性中心,直接生成碳自由基中间体.
在过去几十年中,支持不同假设机理的研究结果纷纷被报道.一些实验结果显示P450催化的脱烷基反应中涉及Cα-H键氧化过程的动力学同位素效应相对较小[6,9-12],是一个Cα的去质子化过程,Kurebayashi[13]进而指出环己胺的脱氨基反应同样显示出较小的动力学同位素效应;同时,在P450及其同功酶的催化下,针对对位取代的苯胺脱甲基反应中线性自由能关系的研究结果得到一个较大的负Hammettρ值,并且反应速率常数与底物的氧化还原势有关[14-16].这些研究结果都支持了反应是涉及SET机理的.然而Jurva[17]等人针对P450催化下的环丙基胺氧化反应的模型电化学-质谱的研究结果显示,反应中的氨基阳离子自由基中间体并不是必须的;李春森[18]等人针对P450催化苯胺羟基化反应和王永[19]等人针对P450催化N,N-二甲基苯胺脱甲基反应的理论计算结果都无一例外地支持了HAT机理.
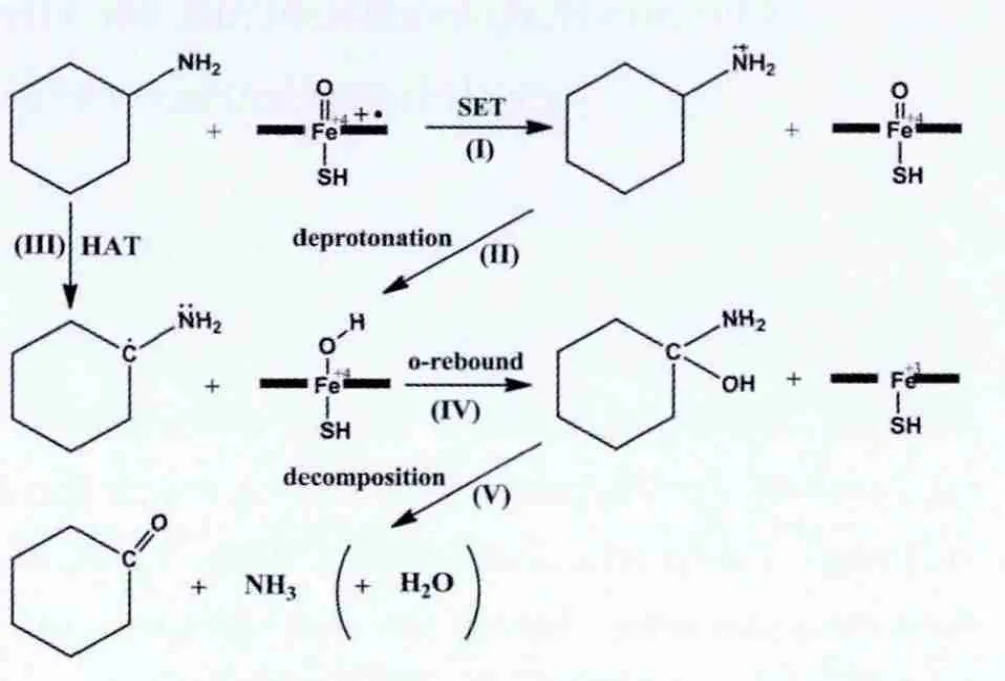
图1 P450催化环己胺脱氨基反应的两种假设机理Fig.1 Two hypothetical mechanisms of the deamination of cyclohexylamine catalyzed by P450
为了揭示由P450催化的环己胺脱氨基反应中涉及的反应机理细节,我们进行了一系列的密度泛函理论(DFT)计算的研究,并以DFT计算结果为基础进行了相关的动力学同位素效应计算,结果显示反应中Cα-H键氧化的过程是一个氢原子传递过程,从理论计算的角度很好地支持了HAT机理,计算结果还显示反应最后一步分解过程是在一个水分子的辅助下在非酶环境中进行的.
2 计算模型与方法
为了通过密度泛函理论计算揭示P450催化环己胺脱氨基反应的机理细节,我们使用了Fe4+O2-(C20N4H12)-(SH)-化 合 物[19-22]作 为 P450氧 化单体Cpd I的模型,使用环己胺(C6H13N)作为底物.相关的DFT计算通过使用Gaussian 09[23]软件包来实现,其中涉及混合泛函B3LYP[24]以及两个基组:一个是 LACVP(Fe)/6-31G**(H,C,N,O,S),简称为B1基组,用来对过渡态和其它稳定点进行非对称性限制的优化;另一个是LACV3P+**(Fe)/6-311+G**(H,C,N,O,S),简称为B2基组,用来进行单点能(E1)和溶剂化校正(E2)的计算.上述计算模型与计算方法已被广泛应用并被证明是十分可靠的[19,22,25-29].
通过振动频率分析,稳定点没有虚频,而过渡态有且只有一个虚频,且该虚频的振动方向是沿着反应坐标方向的.我们还使用了非极性的PCM溶剂化模型(溶剂是介电常数ε=5.697的氯苯)来模拟蛋白质环境下活性中心的极化效应.
为了得到Cα-H键氧化过程的动力学同位素效应(KIE)值,我们还利用通过Gaussian计算得到的频率数据进行了相关的KIE计算,计算主要基于以下两个方程[30],第一个是半经验的Eyring方程,KIE表示为:
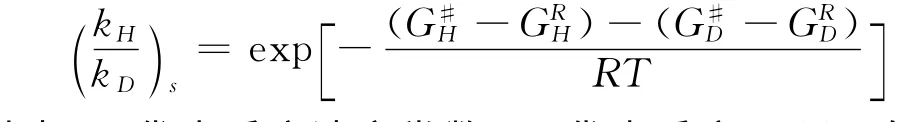
其中,k代表反应速率常数,G代表反应Gibbs自由能,R代表气体常数,T代表绝对温度,下标s代表了该KIE值是半经验值.第二个表达式通过将半经验的 (kH/kD)s与 Wigner量子校正Qcorr因子相乘做了简单的量子化校正,KIE表示为:
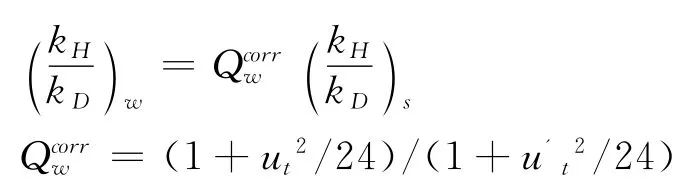
其中ut=hνH/kBT,=hνD'/kBT,ν为过渡态的虚频值,下标w代表了该KIE值为Wigner校正值.
3 结果与讨论
3.1 P450催化环己胺Cα-H键氧化反应
如图2所示,与P450催化的其它反应一样,环己胺Cα-H键氧化反应也是在由两个简并的Cpd I基态引起的不同自旋态上进行的.反应物阶段(RC),在气相环境(E1)和溶剂化环境(E2)下,四重态和二重态是简并的,能差均为0.2kcal/mol.到了过渡态阶段(TS),气相下四重态和二重态的反应能垒分别达到了5.9kcal/mol和5.5 kcal/mol,其能差仍然很小,仅为0.4kcal/mol;而在溶剂化环境中,其反应能垒分别增加到了7.7 kcal/mol和 6.1kcal/mol,能差被扩大至 1.6 kcal/mol.接下来的氧反弹过程是一个无垒过程,生成了初级产物1-羟基-环己胺以及弛豫态的Cpd I,整个过程是一个高放热过程,在气相环境下,四重态和二重态的能量分别下降到-60.0kcal/mol和-60.1kcal/mol,有趣的是,能差仅为0.1kcal/mol,这说明整个反应过程符合Shaik提出的“双态反应”(TSR)机理[21,31-32].

图2 四、二重态下P450催化环己胺Ca-H键氧化反应的能量过程图(kcal/mol).括号外为E1计算水平下的相对能量,括号内为E2计算水平下的相对能量.所有数据都包含了零点能校正.Fig.2 Energy profile(in kcal/mol)for the Cα-H bond oxidation of cyclohexylamine catalyzed by P450on the quartet and doublet states.Relative energies at the E1level are given out of the parentheses and those at the E2level are given in parentheses.All data include ZPE correction.
如图3所示,在过渡态阶段,沿着反应方向,四重态中C-H的距离为1.235Å,H-O的距离为1.369Å,二重态中C-H的距离为1.185Å,H-O的距离为1.472Å,相比之下,四重态的过渡态更加对称,并且由于四重态的C-H距离大于二重态的C-H距离,所以四重态的过渡态相比二重态更“晚”一点.但是,通过对比反应方向上C-H-O的角度可以发现,四重态的C-H-O部分更弯曲,其线性关系并不如二重态的好.
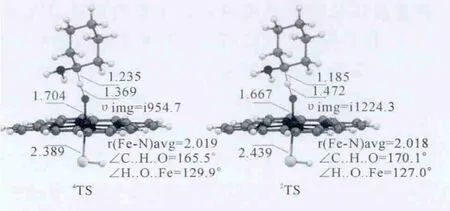
图3 Ca-H键氧化反应过程中DFT优化的四、二重态过渡态构型.键长单位为Å.Fig.3 DFT optimized geometries of the transition state on the quartet and doublet states in the Cα-H bond oxidation process.Bond length is inÅ.
众所周知,自旋密度是分析电子结构最准确也是最直观的方法,因此将P450催化环己胺Cα-H键氧化反应过程中的自旋密度与电荷在表1中列出.在RC阶段,两个自旋态下底物上的自旋均基本为零,而Cpd I的铁-氧部分上积聚了约2个单位的自旋,不同的是,四重态的卟啉-硫氢部分上积聚约1个单位的自旋而在二重态的卟啉-硫氢部分上积聚了约-1个单位的自旋.到了TS阶段,四重态底物上的自旋密度约为0.5,同时二重态底物上的自旋密度约为-0.3;与此同时,Cpd I上卟啉-硫氢部分上的自旋密度也发生了相应的变化,四重态和二重态的自旋密度分别约为0.5和-0.5,这说明TS阶段涉及了电子从底物向Cpd I卟啉-硫氢部分上传递的过程.最后,在PC阶段,底物上的自旋密度又变为了零,而Cpd I上的自旋密度也发生了相应的变化,这同样也说明在无垒的氧反弹过程中也涉及了电子的传递.结合上述的过渡态构型的相关信息,整个Cα-H键氧化反应过程中底物和Cpd I上自旋密度的变化趋势充分说明,在反应的过程中,一个氢原子由底物传递到了Cpd I上,是一个HAT机理.

表1 四、二重态下P450催化环己胺Ca-H键氧化反应的自旋密度与电荷Table 1 Spin densities and charges of the Cα-H bond oxidation of cyclohexylamine on quartet and doublet states catalyzed by P450
表2中,四重态的KIE值为4.9(5.5),略高于二重态的4.1(5.0),较大的 KIE值也与 Miwa和Kurebayshi等人提出的HAT过程具有较大KIE值的特征相吻合,支持了HAT机理.结合上述TS构型相关信息可以看出,KIE值的大小与过渡态的的对称性是成比例的:四重态的C-H距离为1.235Å,明显长于二重态的1.185Å,其过渡态较之二重态也更“晚”,其KIE值也相应的更大.四重态下过渡态的对称性虽明显好于二重态,但其KIE值的差异并不大,我们认为这是由于四重态下过渡态反应方向上C-H-O部分更弯曲,其线性关系并没有二重态好.
2.2 非酶环境下的1-羟基-环己胺分解过程
在P450催化环己胺生成初级产物1-羟基-环己胺之后,接下来的分解过程是在非酶环境中通过一个水分子的辅助进行的.从图4中可以看出,1-羟基-环己胺的分解过程能垒较高,分别达到了17.0kcal/mol和 13.7kcal/mol,而 产 物 阶 段(PC)的能量并没有下降很多,分别为-6.0kcal/mol和-6.9kcal/mol,可以看出整个分解过程放热并不强烈.
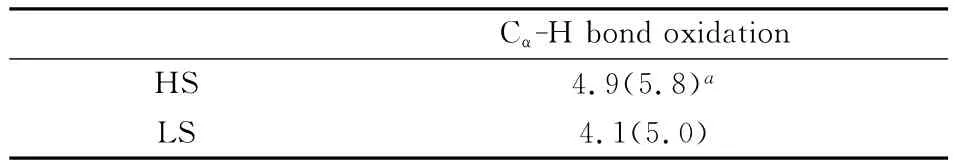
表2 B3LYP/B1计算水平下P450催化环己胺Cα-H键氧化反应过程的内禀动力学同位素效应值Table 2 Calculated intrinsic KIE values for the Cα-H bond oxidation process of cyclohexylamine catalyzed by P450at the B3LYP/B1level
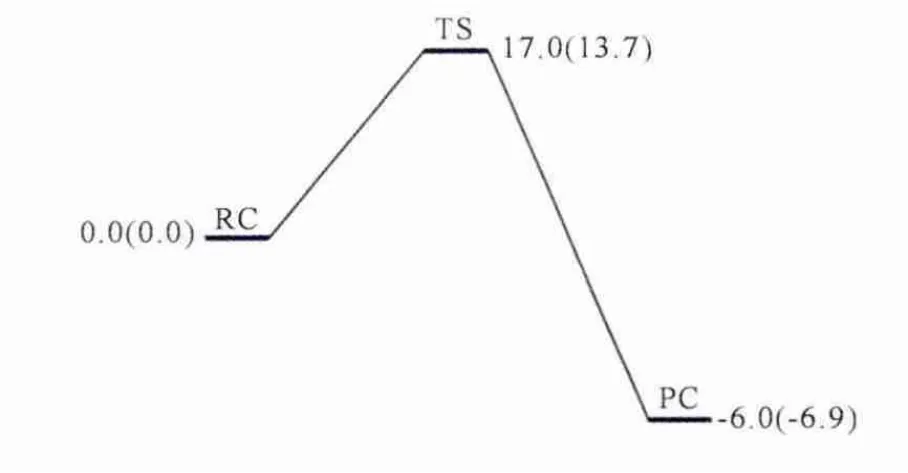
图4 非酶环境下1-羟基-环己胺水分子辅助分解过程的能量过程图(kcal/mol).括号外为E1计算水平下的相对能量,括号内为E2计算水平下的相对能量.所有数据都包含了零点能校正.Fig.4 Energy profile(in kcal/mol)for the decomposition of 1-hydroxycyclohexylamine assisted bya water molecule in nonenzymatic environment.Relative energies at the E1level are given out of the parentheses and those at the E2level are given in parentheses.All data include ZPE correction.
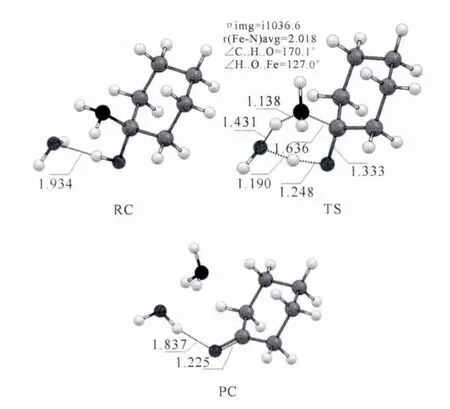
图5 非酶环境下1-羟基-环己胺水分子辅助分解过程中DFT优化的关键中间体构型.键长单位为Å.Fig.5 DFT optimized geometries of the key intermediates in the decomposition process of 1-hydroxycyclohexylamine assisted by one water molecule in the nonenzymatic environment.Bond length is inÅ.
图5给出了1-羟基-环己胺分解过程过渡态的构型信息.水分子在整个分解过程中充当了氢原子传递的“shuttle”,将羟基上的氢原子“传递”到了氨基上,使得1-羟基-环己胺分解为环己酮和氨气.并且从过渡态的构型可以看出,1-羟基-环己胺的分解过程是一个协同的过程.
4 结 论
在P450细胞色素蛋白酶催化的一系列二级胺、三级胺的脱烷基、脱氨基反应中,环己胺的脱氨基反应因其在生物体代谢过程中的重要地位而倍受关注.该反应中涉及Cα-H键氧化过程的机理在一直倍受争议,为了明确其机理细节,本文针对P450催化环己胺脱氨基反应进行了一系列的密度泛函理论(DFT)计算,并在此基础上计算了
Cα-H键氧化过程的动力学同位素效应(KIE).计算结果显示,两个自旋态的Cα-H键氧化反应能垒分别为5.9/5.5kcal/mol,符合Shaik提出的“双态反应”(TSR)机理,并且通过对该反应过程中的过渡态构型以及自旋密度与电荷的分析,证明Cα-
H键氧化过程是一个氢原子传递过程.同时,动力学同位素效应计算结果显示Cα-H键氧化过程有着较大的 KIE值(4.1/4.9),符合 HAT机理的动力学同位素效应特征.此外,通过针对Cα-H键氧化反应的初级产物的分解过程进行了DFT计算,发现通过一个水分子的辅助,在非酶环境中整个分解过程放热并不强烈,并且该过程为一个协同的过程.
[1]Ortiz de Montellano P,Voss J.Substrateoxidation bycytochromeP450enzymes[M].New York:Plenum Publishers,2005:183.
[2]Manchester J I,Dinnocenzo J P,Higgins L A,et al.A new mechanistic probe for cytochrome P450:An application of isotope effect profiles [J].J.Am.Chem.Soc.,1997,119(21):5069.
[3]Karki S B,Dinnocenzo J P,Jones J P,etal.Mechanism of oxidative amine dealkylation of substituted N,N-dimethylanilines by cytochrome P450:application of isotope effect profiles[J].J.Am.Chem.Soc.,1995,117(13):3657.
[4]Karki S B,Dinnocenzo J P.On the mechanism of amine oxidations by P450[J].Xenobiotica,1995,25(7):711.
[5]Dinnocenzo J P,Karki S B,Jones J P.On isotope effects for the cytochrome P450oxidation of substituted N,N-dimethylanilines [J].J.Am.Chem.Soc.,1993,115(16):7111.
[6]Guengerich F P,Macdonald T L.Mechanisms of cytochrome P450catalysis[J].FasebJ.,1990,4(8):2453.
[7]Groves J T.Key elements of the chemistry of cytochrome P450:the oxygen rebound mechanism [J].J.Chem.Educ.,1985,62(11):928.
[8]Groves J T,McClusky G A.Aliphatic hydroxylation via oxygen rebound:oxygen transfer catalyzed by iron[J].J.Am.Chem.Soc.,1976,98(3):859.
[9]Abdelmonem M M.Isotope effects in enzymatic N-demethylation of tertiary amines [J].J.Med.Chem.,1975,18(4):427.
[10]Hollenberg P F,Miwa G T,Walsh J S,etal.Mechanisms of N-demethylation reactions catalyzed by cytochrome P450and peroxidases [J].Drug Metab.Dispos.,1985,13(3):272.
[11]Miwa G T,Garland W A,Hodshon B J,etal.Kinetic isotope effects in cytochrome P450-catalyzed oxidation reactions:intermolecular and intramolecular deuterium isotope effects during the N-demethylation of N,N-dimethylphentermine [J].J.Biol.Chem.,1980,255(13):6049.
[12]Miwa G T,Walsh J S,Kedderis G L,etal.The use of intramolecular isotope effects to distinguish between deprotonation and hydrogen atom abstraction mechanisms in cytochrome P450-catalyzed and peroxidase-catalyzed N-demethylation reactions[J].J.Biol.Chem.,1983,258(23):4445.
[13]Kurebayashi H.Kinetic deuterium isotope effects on deamination and N-hydroxylation of cyclohexylamine by rabbit liver microsomes [J].Arch.Biochem.Biophys.,1989,270(1):320.
[14]Burka L T,Guengerich F P,Willard R J,etal.Mechanism of cytochrome P450catalysis:mechanism of N-dealkylation and amine oxide deoxygenation[J].J.Am.Chem.Soc.,1985,107 (8):2549.
[15]Galliani G,Nali M,Rindone B,etal.The rate of N-demethylation of N,N-dimethylanilines and N-methylanilines by rat liver microsomes is related to their 1st ionization potential,their lipophilicity and to a steric bulk factor[J].Xenobiotica,1986,16(6):511.
[16]Galliani G,Rindone B,Dagnino G,etal.Structure reactivity relationships in the microsomal oxidation of tertiary amines[J].Eur.J.DrugMetab.Ph.,1984,9(4):289.
[17]Jurva U,Bissel P,Isin E M,etal.Model electrochemical-mass spectrometric studies of the cytochrome P450-catalyzed oxidations of cyclic tertiary allylamines[J].J.Am.Chem.Soc.,2005,127(35):12368.
[18]Li C S,Wu W,Kumar D,etal.Kinetic isotope effect is a sensitive probe of spin state reactivity in C-H hydroxylation of N,N-dimethylaniline by cytochrome P450[J].J.Am.Chem.Soc.,2006,128(2):394.
[19]Wang Y,Kumar D,Yang C L,etal.Theoretical study of N-demethylation of substituted N,N-dimethylanilines by cytochrome P450:The mechanistic significance of kinetic isotope effect profiles[J].J.Phys.Chem.B,2007,111(26):7700.
[20]Shaik S,Kumar D,de Visser S P,etal.Theoretical perspective on the structure and mechanism of cytochrome P450enzymes[J].Chem.Rev.,2005,105(6):2279.
[21]Ogliaro F,Harris N,Cohen S,etal.A model"rebound"mechanism of hydroxylation by cytochrome P450:Stepwise and effectively concerted pathways,and their reactivity patterns[J].J.Am.Chem.Soc.,2000,122(37):8977.
[22]Wang Y,Yang C L,Wang H M,etal.A new mechanism for ethanol oxidation mediated by cytochrome P450 2E1:Bulk polarity of the active site makes a difference [J].ChemBioChem,2007,8(3):277.
[23]Frisch M J,Trucks G W,Schlegel H B,Scuseria G E,Robb M A,Cheeseman J R,Scalmani G,Barone V,Mennucci B,Petersson G A,Nakatsuji H,Caricato M,Li X,Hratchian H P,Izmaylov A F,Bloino J,Zheng G,Sonnenberg J L,Hada M,Ehara M,Toyota K,Fukuda R,Hasegawa J,Ishida M,Nakajima T,Honda Y,Kitao O,Nakai H,Vreven T,Montgomery J A,Jr,Peralta J E,Ogliaro F,Bearpark M,Heyd J J,Brothers E,Kudin K N,Staroverov V N,Keith T,Kobayashi R,Normand J,Raghavachari K,Rendell A,Burant J C,Iyengar S S,Tomasi J,Cossi M,Rega N,Millam J M,Klene M,Knox J E,Cross J B,Bakken V,Adamo C,Jaramillo J,Gomperts R,Stratmann R E,Yazyev O,Austin A J,Cammi R,Pomelli C,Ochterski J W,Martin R L,Morokuma K,Zakrzewski V G,Voth G A,Salvador P,Dannenberg J J,Dapprich S,Daniels A D,FarkasÖ,Foresman J B,Ortiz J V,Cioslowski J,Fox D J,Gaussian09,revision C.01.Wallingford.CT:Gaussian.Inc.,2010.
[24]Lee C T,Yang W T,Parr R G.Development of the colle-salvetti correlation energy formula into a functional of the electron density[J].Phys.Rev.B,1988,37(2):785.
[25]Chen H,Song J S,Lai W Z,etal.Multiple low-lying states for compound I of P450(cam)and chloroperoxidase revealed from multireference ab initio QM/MM calculations[J].J.Chem.TheoryComput.,2010,6(3):940.
[26]Lai W Z,Chen H,Cohen S,etal.Will P450(cam)hydroxylate or desaturate alkanes?QM and QM/MM studies[J].J.Phys.Chem.Lett.,2011,2(17):2229.
[27]Lai W Z,Chen H,Cho K B,etal.Effects of substrate,protein environment,and proximal ligand mutation on compound I and compound 0of chloroperoxidase[J].J.Phys.Chem.A,2009,113(43):11763.
[28]Li D M,Wang Y,Han K L.Recent density functional theory model calculations of drug metabolism by cytochrome P450 [J].Coord.Chem.Rev.,2012,256(11-12):1137.
[29]Wang Y,Li D M,Han K L,etal.An acyl group makes a difference in the reactivity patterns of cytochrome P450catalyzed N-demethylation of substituted N,N-dimethylbenzamides-high spin selective reactions[J].J.Phys.Chem.B,2010,114 (8):2964.
[30]Melander L,Saunders W H,Reactionratesofisotopicmolecules[M].Malabar,Fla.:RE Krieger Pub.Co.,1987.
[31]Schroder D,Shaik S,Schwarz H.Two-state reactivity as a new concept in organometallic chemistry[J].AccountsChem.Res.,2000,33(3):139.
[32]Shaik S,Filatov M,Schroder D,etal.Electronic structure makes a difference:Cytochrome P-450 mediated hydroxylations of hydrocarbons as a twostate reactivity paradigm [J].Chem.-Eur.J.,1998,4(2):193.
猜你喜欢
吉林大学学报(理学版)(2024年5期)2024-01-01 00:00:00
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
云南化工(2021年6期)2021-12-21 07:30:56
大学化学(2021年8期)2021-09-26 10:51:16
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
科学(2020年2期)2020-08-24 07:57:00
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
生物技术通报(2015年1期)2015-04-10 16:15:19
无机化学学报(2014年12期)2014-02-28 17:34:01
