
吡咯烷酮对Aun(n=2~10)团簇保护机制的第一性原理研究
2014-07-13 03:39:12高艳蓉余盛萍赵志刚杨明理
原子与分子物理学报 2014年3期
高艳蓉,余盛萍,赵志刚,杨明理
(1.西南民族大学化学与环境保护工程学院,成都610041;2.四川大学原子分子研究所,成都610065)
1 引 言
Au是化学性质最稳定的金属之一,在一般条件下没 有 催 化 活 性[1].1987 年Haruta等 人[2]发 现TiO2或Fe2O3载体上的Au团簇在低温下能催化CO氧化反应,打破了Au没有催化活性的传统观念,引起了对Au团簇催化特性的研究兴趣.随后的研究发现[3-5],Au团簇具有较高活性,这有利于其催化作用,但也使其应用条件更为苛刻.较小的Au团簇之间具有聚集形成较大团簇的趋势,会降低表面能,增大团簇结构的稳定性,同时也导致其活性降低.为阻止团簇之间的聚集作用,近年来发展了利用有机配体保护团簇的方法.在团簇表面预吸附有机配体,利用空间效应阻碍其它团簇靠近.同时,通过调节配体与团簇之间的作用,保持甚至加强团簇的活性.poly(N-vinyl-2-pyrrolidone)(PVP)是一种常用的保护配体.最近,Tsunoyama 等人[6-8]提出了PVP与Aun之间的作用模型,认为PVP在保护Au的同时,与Au团簇之间存在电子耦合效应,帮助Aun保持催化活性.但是,尚无理论计算支持这一模型.第一性原理计算是研究团簇及其吸附物质之间相互作用的有效工具,本文利用密度泛函理论计算研究Aun(n=2-10)与PVP之间形成的吸附产物的几何结构和电子结构,分析PVP吸附对Aun结构和性质的影响,为进一步研究配体保护下Aun催化反应机理奠定基础.
2 计算方法
鉴于对Au团簇的结构已有大量的理论研究,我们收集文献中关于Aun(n=2-10)[9]结构参数,在TPSS/def2-TZVP[10]水平下进行重新优化.PVP分子结构复杂,为了简化计算,用H 代替PVP中的 基团.为与PVP区别,配体简称PRD.在TPSS/def2-TZVP 水平上对大量Aun:PRD(n=2-10)起始构型进行优化和遴选,得到较为稳定的吸附产物构型.并在相同水平下计算了最稳定构型的振动频率以确定其为势能面上的极小点.为了比较,同时用另外两种泛函PBE[15]和BLYP[16,17]在相同的基组下进行了类似计算.
所有计算使用Turbomole程序包完成[11].有效核电势采用了 Wood-Boring 相对论校正(MWB)[12]来描述60核电子,而价电子5s、5p、5d和6s采用了三重分裂的价电子基组加增强的极化函数,对C、N、O 和H 也采用了相同的def2-TZVP基组.计算得到Au2中Au—Au键长为2.521Å,与实验值2.47Å[13]相近.并与用PW91的理论计算值2.528Å[14]吻合较好.
3 结果与讨论
图1列出了优化得到的吸附产物Aun∶PRD的较为稳定的构型.对于n≥4,只列出最为稳定的三个构型.在图中标出了所有最稳定构型的C—O键和Au—O 键键长,括号内是相对能量.命名中间的数字表示Au原子数目,最后一个字母表示异构体的能量顺序.例如,D3b 代表是由PRD 吸附到Au3得到的次稳定的Au3∶PRD.在图1所示结构中,所有吸附均发生于PRD 中的O 和Aun中的单个原子之间.我们的计算中包含了其它吸附方式,如PRD 中其它原子参与吸附、Aun中的桥位或者多位吸附等,但获得的吸附产物能量明显高于图1中的O-Au单吸附方式.Au2只有一种异构体,也仅有一种较稳定吸附产物D2a.Au3有3 种异构体,PRD 吸附到三角形构型上得到最稳定的吸附产物D3a,比D3b稳定0.12eV.D4a由PRD 吸附到Y 型的Au4上得到,其能量分别比D4b和D4c的低0.27和0.69eV.PRD 吸附在Au5梯形异构体的顶点原子上,形成D5a,其能量比D5b和D5c的分别低0.03 和0.22eV.D6a中,PRD 吸附在Au6的平面D3h构型的顶点原子上,比D6b和D6c分别稳定0.42和0.94eV.注意到D6c中的Au6为立体构型,这是在较稳定吸附产物中首次出现3D 的Aun构型.对于n=7-10,PRD 的吸附方式与较小团簇相似,PRD 中的O 原子倾向于首先吸附到Aun中的顶点原子,其次是边缘原子.在绝大部分吸附产物中,Aun部分保持平面构型.PRD 部分具有平面结构,吸附后基本保持不变,由于分子可以绕Au—O 键转动,吸附产物中,PRD 并不与Aun保持在同一平面.我们注意到D10c也具有3D 结构,与D10a的能量差为0.14eV,远小于另一个具有3D Aun构型的D6c 与D6a 的能量差(0.94eV).
我们定义吸附能为
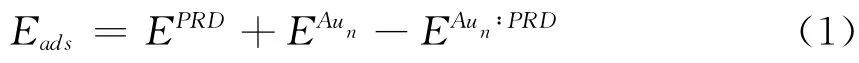
这 里 的EPRD,EAun ,和EAun∶PRD分 别 表 示PRD,Aun团簇,和Aun∶PRD 的能量.图2列出了图1中最稳定吸附产物的吸附能随Aun中原子的数目而变化.对于n=1-10,Eads 在0.62-1.35eV 之间变化,表明PRD 能够在Aun上形成较为稳定的吸附产物.其中,D4a的吸附能最大,为1.35eV,主要原因是其吸附前的Aun构型(Y型)能量较高,Au4的最稳定构型为菱形.D5a和D6a吸附能最小,均为0.62eV.从n=6开始,具有奇数Au原子数的Aun:PRD 的吸附能明显大于其相邻的偶数Aun∶PRD,这与文献中奇数Au团簇比偶数团簇活性较高的结论一致.

图1 Aun∶PRD(n=2-10)较稳定异构体的结构,键长单位:Å.括号里是相对能量,单位:eV.Fig.1 Optimized geometries of Aun∶PRD(n=2-10)isomers.All bond lengths are inÅ.The values in parentheses are the relative energies(in eV).
为了分析吸附过程导致的Au团簇与保护配体之间的电子效应,我们计算了最稳定吸附产物中各原子的净电荷.PRD吸附导致Aun中吸附位附近的原子的净电荷发生较为明显的变化,其中处于吸附位的Au带负电荷,其周围Au原子带正电荷.表1给出了在TPSS/def2-TZVP水平下Au部分的总净电荷,由Aun中各原子的净电荷求和得到.总净电荷在-0.05e到-0.11e之间变化,表明有少量的电子从PRD转移到Aun.可见,PRD 的作用不仅仅限于吸附在Aun表面阻碍其它Aun靠近,而且部分地改变了Aun的电子结构,可能会对其活性产生部分影响.PRD的这种供电子效应有利于Aun作为电子供体参与活化O2的反应.但是,由于PRD 与Aun之间的电荷转移量较小,带来的吸附协同效应并不显著.表1同时给出了奇数团簇吸附产物中的自旋密度分布,绝大部分自旋仍然集中于Aun部分,仅有少量(<7%)分布于PRD.可见PRD 吸附对Aun电子性质的影响有限.

图2 PRD 在Aun(n=2-10)上的吸附能Eads.单位:eV.Fig.2 Adsorption energies EPRDads (in eV)of PRD on Aun(n=2-10).

表1 最稳定Aun∶PRD(n=2-10)复合物中的电荷和自旋密度分布.Table 1 Net charge and spin density of the most stable Aun∶PRD(n=2-10)complexes.

图3 Au6(上)和D6a(下)的HOMO 和LUMO 轨道Fig.3 HOMO and LUMO of the most stable Au6(upper)and D6a(lower).
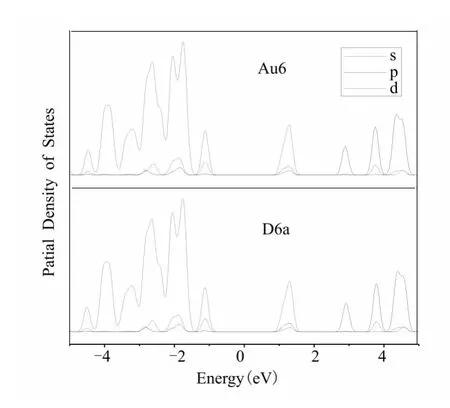
图4 Au6 和D6a的PDOS.红,蓝,绿线分别表示s,p,d态.Fig.4 PDOS of Au6and D6a.The s-,p-and dtype contributions are in red,blue and green,respectively.
我们进一步分析PRD 吸附前后体系的Kohn-Sham 能级的变化.图3和图4分别比较了Au6在吸附生成D6a前后的HOMO 和LUMO 构成和态密度分析,图1所示其它各稳定吸附产物具有类似结果.与Au6相近,D6a的HOMO 和LUMO 主要都分布在Au上,仅有极少量分布在与Aun相连的O 原子上.虽然D6a的LUMO 与Au6的LUMO 不同,但这并不影响Au6的催化活性,因为Aun活化O2时作为供电子基团出现,其催化活性主要由其HOMO 决定.Au6和D6a具有相似的态密度分布.HOMO-LUMO 能级差基本保持不变,Fermi能级附近的占据轨道主要由d和s态贡献,非占据轨道主要由s和p态贡献.PRD 吸附对Aun的态密度影响极小.
4 结 论
在TPSS/def2-TZVP 水平下,计算研究了配体PRD 对Aun(n=2-10)团簇的保护机制.研究了一系列Au团簇与PRD 之间的相互作用,找到它们之间的作用模式,得到以下结论:(1)PRD 倾向吸附于Aun(n=2-10)团簇的顶点,PRD 中的O 与Au相互间形成较弱的Au-O 键,其本质是物理吸附;(2)团簇尺寸对吸附作用有较大影响,从n=6开始,具有奇数原子数的Aun∶PRD 的吸附能明显大于其相邻的偶数团簇.(3)在PRD 和Aun之间存在少量的电荷转移,PRD 作为电子供体,Au团簇为电子受体,表明PRD 的存在不仅能阻碍Aun的聚集,而且部分地改变了Aun的电子结构,可能会对其活性产生影响.我们的计算支持Tsunoyama等人[6-8]的模型,但是我们发现PRD对Aun的活性的影响较小,表现在吸附仅带来Aun电子性质的较小变化.这正是PRD 作为Aun保护基的结构基础.
[1] Hammer B,Nørskov J K.Why gold is the noblest of all the metals[J].Nature,1995,376:238.
[2] Haruta M,Kobayashi T,Sano H,et al.Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0℃[J].Chem.Lett.,1987,405.
[3] Kim T S,Stiehl J D,Reeves C T,et al.Cryogenic CO oxidation on TiO2-supported gold nanoclusters precovered with atomic oxygen[J].J.Am.Chem.Soc.2003,125:2018.
[4] Deng X H,Li P,Cheng Z,et al.Geometries and electronic structures of gold cluster from density functional theory[J].J.At.Mol.Phys.,2007,24:347(in Chinese)[邓小辉,李萍,程长,等.金团簇结构和电子性质的密度泛函研究[J].原子与分子物理学报,2007,24:347]
[5] Mao L P,Tian D X,Guo X Y.First-principles study of H2O and O2adsorption on Au38cluster[J].J.At.Mol.Phys.,2010,27:69(in Chinese)[毛丽萍,田东旭,郭向云.Au38团簇上小分子H2O 和O2共吸附的第一性原理研究[J].原子与分子物理学报,2010,27:69]
[6] Tsunoyama H,Ichikuni N,Sakurai H,et al.Effect of electronic structures of Au clusters stabilized by poly(N-vinyl-2-pyrrolidone)on aerobic oxidation catalysis[J].J.Am.Chem.Soc.,2009,131:7086.
[7] Tsunoyama H,Sakurai H,Negishi Y,et al.Sizespecific catalytic activity of polymer-stabilized gold nanoclusters for aerobic alcohol oxidation in water[J].J.Am.Chem.Soc.,2005,127:9374.
[8] Tsunoyama H,Sakurai H,Tsukuda T.Size effect on the catalysis of gold clusters dispersed in water for aerobic oxidation of alcohol[J].Chem.Phys.Lett.,2006,429:528.
[9] Assadollahzadeh B,Schwerdtfeger P.A systematic search for minimum structures of small gold clusters Aun(n=2-20)and their electronic properties[J].J.Chem.Phys.,2009,131:064306.
[10] Tao J,Perdew J P,Staroverov V N,et al.Climb-ing the density dunctional ladder:nonempirical meta-generalized gradient approximation designed for molecules and solids[J].Phys.Rev.Lett.,2003,91:46401.
[11] TURBOMOLE V5-9-1.University of Karlsruhe 2007.
[12] Wood J H,Boring A M.Improved Pauli Hamiltonian for local-potential problems[J].Phys.Rev.B.,1978,18:2701.
[13] Huber K P,Herzberg G.Molecular Spectra and Molecular Structure,Van Nostrand Reinhold,New York,1979.
[14] Mills G,Gordon M S,Metiu H.The adsorption of molecular oxygen on neutral and negative Aunclus-ters(n=2-5)[J].Chem.Phys.Lett.,2002,359:493.
[15] Perdew J P,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Phys.Rev.Lett.,1996,77:3865.
[16] Becke A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Phys.Rev.A.,1988,38:3098.
[17] Lee C L,Yang W,Parr R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].Phys.Rev.B.,1988,37:785.
猜你喜欢
物理通报(2024年4期)2024-04-09 12:41:28
中学生数理化·中考版(2021年10期)2021-11-22 07:26:40
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27 03:07:02
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
新高考·高一物理(2015年6期)2015-09-28 20:10:57
航天返回与遥感(2014年4期)2014-07-31 17:47:47
无机化学学报(2014年6期)2014-02-28 17:32:06
