
LC-MS/MS法测定人血浆中羟基红花黄色素A的浓度
2014-05-17 03:03李长印储继红臧雨馨戴国梁邹建东居文政
中国药理学通报 2014年10期
李长印,储继红,张 军,臧雨馨,戴国梁,邹建东,居文政
(1.南京中医药大学附属医院临床药理实验室,江苏南京 210029;2.南京中医药大学药学院,江苏 南京 210002)
目前已报道的人血浆中QA含量的测定方法主要有 HPLC法[5-6]和 LC-MS/MS法[7]。HPLC法由于检测灵敏度较低,无法保证完全检测到低给药剂量下各个时点的人体QA血药浓度,从而保障药代动力学的顺利开展;同时由于检测波长下杂质干扰的影响,实现化合物的完全分离和准确定量往往采用梯度洗脱,因此需要更长的分析时间,不利于药代动力学研究中样品的批量分析。而LC-MS/MS法具有更高的检测灵敏度和更小的杂质信号干扰,因此更适用于测定人血浆中QA的浓度。但在已报道的LC-MS/MS方法中[7],研究者采用 SPE方法对血浆样品进行前处理,在进样前必须进行吹干和复溶等操作,具有不利于高通量样品分析的缺点。同时,上述报道中LC-MS/MS方法的建立是为了研究单剂量口服QA制剂的药代动力学研究,因此线性范围偏窄,未进行稀释比实验等方面的考察。而开展系统的QA人体药动学研究一般涉及高、中、低3个剂量组的血药浓度测定,在保证低剂量组血药浓度检测灵敏度的前提下,高剂量组的血药浓度往往会超出定量上限,因此有必要进行稀释比实验等相关考察。基于此,本研究拟对已报道的测定方法进行系统优化,并对方法进行全面的方法学验证,以建立一种灵敏、简便、快速、可靠的QA人体血药浓度测定方法,用于人体药代动力学的系统研究。
1 材料
1.1 药品与试剂 羟基红花黄色素A(QA)对照品(中国食品药品检定研究院,批号111637-201106,含量92.5%,结构如Fig 1A所示);异鼠李素-3-O-新橙皮苷(SLS)对照品(内标,中国食品药品检定研究院,批号111571-201205,含量93.2%,结构式见Fig 1B);醋酸铵(Tedia Company Inc.,HPLC级,批号20266-0500);甲醇(Merck Company,HPLC级);超纯水由Millipore Milli-Q Advantage A10超纯水机制备;其余试剂为分析纯。注射用QA由广州悦康生物制药有限公司提供,25 mg/瓶,临用前用0.9%氯化钠注射液250 ml稀释,静脉滴注,输液泵流速4 ml/min。人空白血浆源自江苏省血液中心。
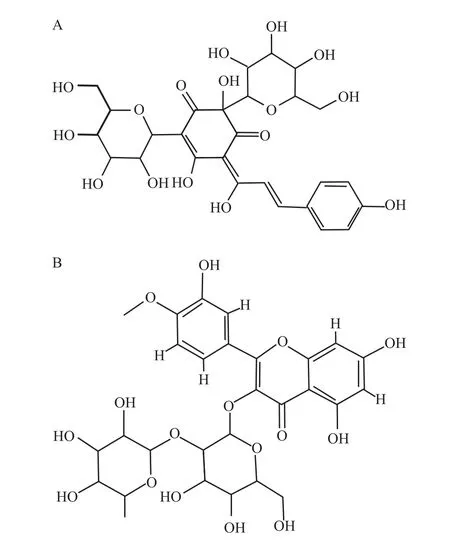
Fig 1 Chem ical structures of QA(A)and internal standard(SLS,B)
1.2 实验仪器 LC(Agilent)-(AB)API4000 MS/MS液质联用仪,含Pump Agilent 1260G1312A、Column Oven Agilent1260G1316A、Auto Sampler Agilent 1260G1367E和 Mass spectrometer API4000,LC-MS/MS系统由Analyst1.6 Software控制;CPA225D电子天平(德国Sartorius公司);高速低温离心机(Thermo Sorvall Legend Micro 17R,PT021);WH2微型旋涡混合仪(上海沪西分析仪器厂);SPE小柱(waters Oasis@HLB 3cc(60 mg)Extraction Cartridges)。
2 方法与结果
2.1 色谱与质谱条件 色谱条件:Agilent ZORBAX SB C18色谱柱(4.6 mm×150 mm,5μm),Agilent ZORAX SB-C8预柱(2.1×12.5 mm,5μm),流动相组成为 0.2 mol·L-1乙酸铵水溶液/甲醇(30/70),流速400μl/min,柱温35℃,分析 5 min,进样体积5μl。
质谱条件:ESI离子源,负离子模式检测,扫描方式为多反应检测(MRM),QA的检测目标离子对为 m/z 611.131/490.900,DP为 -125 V,CE为-34 eV,SLS的检测目标离子对为 m/z 623.032/298.800,DP为-150 V,CE为 -3462ev。其他主要质谱参数如下:Dwell Time 150 ms,ED-10V,CXP-11V,碰撞气(CAD)10,气帘气(CUR)25,辅助气 1(GS1)50,辅助气 2(GS2)55,电喷雾电压(IS)-4500,离子源温度(TEM)400。
2.2 标准溶液的配制
2.2.1 QA对照品储备溶液的配制 精密称取QA对照品11.31 mg(相当于含QA 10.46 mg)于10 ml容量瓶中,以甲醇溶解、定容,混匀即得浓度为1.046 g·L-1的QA标准曲线储备液,于4℃冰箱中保存备用。临用前用甲醇将其逐级稀释得到含QA浓度分别为 0.1714、0.3428、0.857、2.143、5.356、13.39、33.48、83.69 mg·L-1的系列标准曲线工作液。
精密称取QA对照品11.55 mg(相当于含QA 10.68 mg)于10 ml容量瓶中,以甲醇溶解、定容,混匀即得浓度为1.068 g·L-1的QA储备液,于4℃冰箱中保存备用。临用前用甲醇将其逐级稀释得到含 QA浓度分别为0.3297、2.968、71.23 mg·L-1的系列质控工作液。
2.2.2 内标SLS对照品储备溶液的配制 精确称取异鼠李素-3-O-新橙皮苷(SLS)对照品10.66 mg(相当于含SLS 9.935 mg)于10 ml容量瓶中,用甲醇溶解定容,得到浓度为0.9935 g·L-1的内标储备液;将此溶液稀释500倍,即得浓度为1.987 mg·L-1的内标工作液。储备液和工作液均于4℃冰箱中保存备用。
2.3 血浆样品处理
2.3.1 受试者含药血浆样品处理 取血浆400μl于1.5 ml EP管中,依次加入浓度为1.987 mg·L-1的内标工作液20μl、0.2 mol·L-1乙酸铵水溶液400μl,涡旋30 s混匀,12 000×g×5 min,4℃离心,吸取上清液750μl上样已活化的SPE固相小柱,上样完毕后用2 ml水洗除杂,最后用70%甲醇水1 ml洗脱,洗脱液涡旋混匀后进行LC-MS/MS分析。空白血浆样品除了未加内标之外,其余样品处理方法相同。
2.3.2 血浆标准曲线样品的配制与处理 取空白血浆400μl于1.5 ml EP管中,分别加入不同浓度的系列标准曲线工作液,涡旋混匀,即得含QA浓度分别为 8.570、17.14、42.85、107.1、267.8、669.6、1674、4185μg·L-1的系列血浆标准曲线样品,后续处理过程同“2.3.1受试者含药血浆样品处理”项下要求。
2.3.3 血浆质控样品的配制与处理 取空白血浆400μl于1.5 ml EP管中,分别加入不同浓度的系列质控工作液,涡旋混匀,即得含QA浓度分别为16.49、148.4和3561μg·L-1的系列血浆质控样品,后续处理过程同“2.3.1受试者含药血浆样品处理”项下要求。
2.4 分析方法的验证
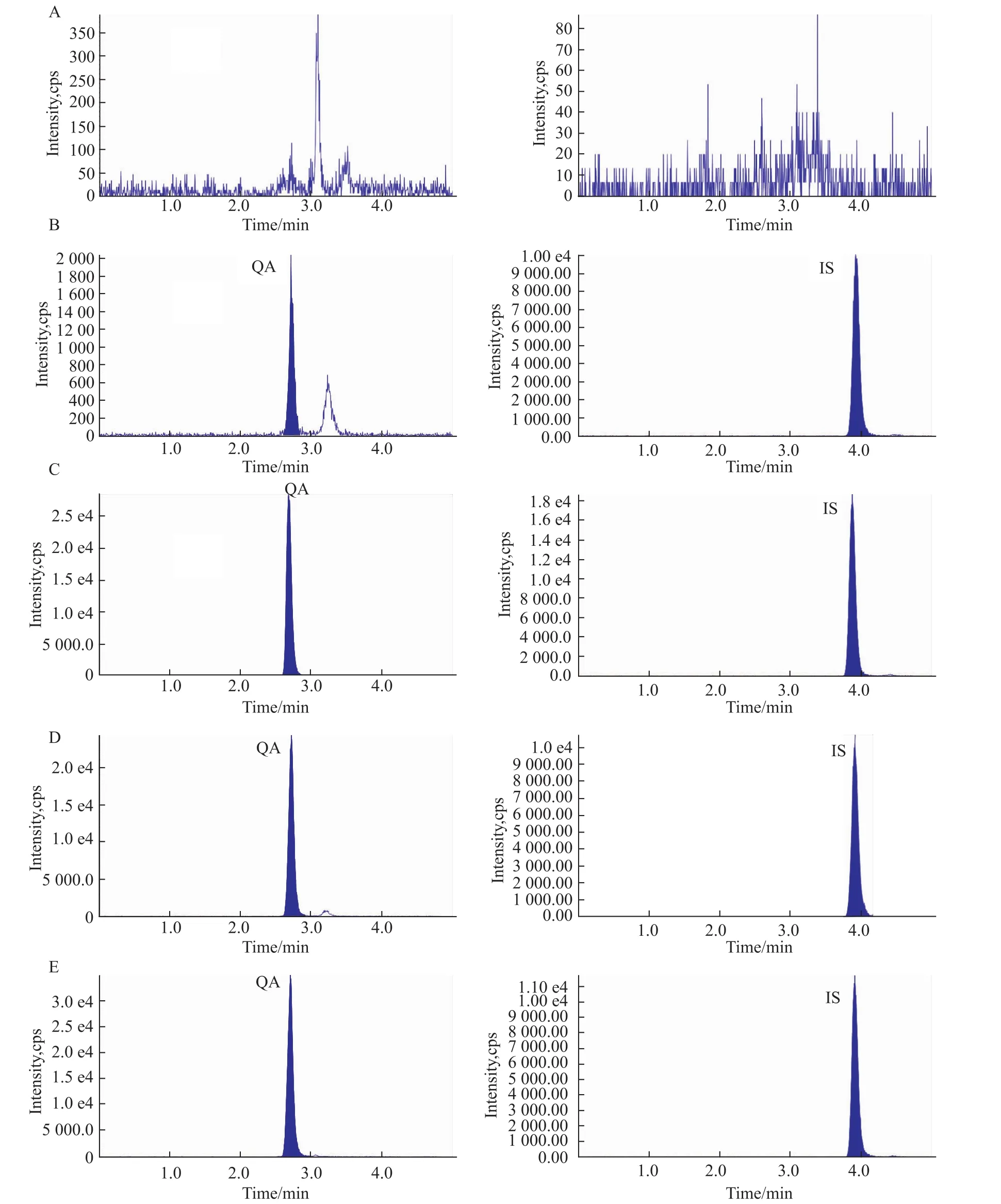
Fig 2 Typical LC-MS/MS chromatograms of(A)blank human plasma;(B)QA spiked plasma sample(8.57μg·L-1)for LLOQ;(C)methanol solution of standard m ixture for QA(59.36μg·L-1)and IS(99.35μg·L-1);(D)QA spiked plasma samp le(148.4μg·L-1)for quality control;(E)p lasma sample obtained from a volunteer at 5 h after drip intravenous infusion of QA injection containing 25 mg of QA
2.4.1 方法的特异性 Fig 2为6份不同来源人空白血浆、定量下限(LLOQ)血浆样品、标准品溶液、质控中浓度血浆样品和受试者静滴QA后血浆样品的特征色谱图。结果表明,在选定的LC-MS/MS条件下,血浆样品中QA和内标SLS的保留时间分别在2.7 min和3.9 min左右,空白血浆中的内源性物质和代谢物等不影响QA的准确测定。
2.4.2 介质效应 按“2.3.1血浆样品处理”项处理6种不同来源的空白血浆,获得其相应的空白血浆洗脱液。分别精密吸取QA低、中、高质控工作液各20μl,向其中分别依次加入内标工作液20μl、上述空白血浆洗脱液960μl,涡旋混匀,吸取5μl进样分析。每种来源的空白血浆均需配制低、中、高质控浓度样品,每个浓度平行3个样本。记录色谱图、QA峰面积As和内标峰面积Ai。计算6种不同来源血浆样品同一浓度的(n=3)和所有浓度的
分别精密吸取QA低、中、高质控工作液各20 μl,向其中分别依次加入内标工作液20μl、甲醇960μl,涡旋混匀,吸取5μl进样分析。每个浓度平行3个样本。记录色谱图、QA峰面积As和内标峰面积Ai。计算同一浓度样品的(n=3)和所有样品的
按下列公式计算介质效应:QA的介质效应ME100%,内标SLS的介质效应MEi×100%。QA低、中、高3个浓度血浆质控样本的平均介质效应分别为116.62%、119.06%和115.72%,RSD分别为3.27%、3.42%和4.93%,内标平均介质为78.97%,RSD分别为1.53%。结果表明,该方法中QA及内标的介质效应精密且可重现。
2.4.3 标准曲线线性、稀释比考察和定量下限 按照“2.3.2血浆标准曲线样品的配制与处理”项配制处理并测定血浆标准曲线样品,记录色谱图、QA峰面积As和内标峰面积Ai。以QA血药浓度C为X轴,以 As与 Ai的峰面积比值 f(f=As/Ai)为 Y轴进行权重回归(权重系数为1/C2),得标准曲线回归方程为Y=(0.0124±0.0017)X+(0.0269±0.0051)(n=9),相关系数r在0.9949~0.9992之间。结果表明:血浆中QA浓度在8.570~4185μg·L-1范围内,浓度与峰面积的比值线性关系良好。同时,稀释比试验结果表明,浓度超出定量上限6.25倍(即26 150μg·L-1)以下的QA血浆样品经空白血浆稀释6.25倍后,实测浓度的准确度为88.03%~90.97%,RSD为1.73% ~5.09%,即通过稀释可以实现高浓度血浆样品药物浓度的准确测定。
按照“2.3.2血浆标准曲线样品的配制与处理”项平行配制处理5份血浆标准曲线最低点样品并进行测定。记录色谱图、QA峰面积As和内标峰面积Ai。计算 As和 Ai的比值 y(y=As/Ai),将 f值代入随行标准曲线求得实测浓度,进而计算其准确度和RSD(n=5)。LLOQ典型色谱图见 Fig 2。结果表明:本方法中QA的LLOQ为8.570μg·L-1,实测浓度的准确度为97.59%,RSD为4.45%。
2.4.4 精密度和准确度考察 按“2.3.3血浆质控样本的配制和处理”项配制处理并测定QA低、中、高3种浓度的血浆质控样本,每个浓度制备5个样本,连续制备并测定3批。记录色谱图、QA峰面积As和内标峰面积Ai。计算As和Ai的比值 f(f=As/Ai),将f值代入随行标准曲线求得实测浓度及其准确度。批内和批间精密度分别用批内(n=5)和3批间(n=15)的浓度实测值的RSD值表示。考察结果如Tab 1所示,结果表明,该方法的批内和批间精密度和准确度良好。
2.4.5 提取回收率考察 按“2.3.3血浆质控样本的配制和处理”项配制处理并测定QA低、中、高3种浓度的血浆质控样本,每个浓度平行3个样本。记录色谱图、QA峰面积(As)B和内标峰面积(Ai)B。按“2.3.1血浆样品的处理”项处理获得空白血浆洗脱液若干,用于配制未经提取含药血浆样品。分别精密吸取QA低、中、高质控工作液各20μl,向其中分别依次加入内标工作液20μl、空白血浆洗脱液960μl,涡旋混匀,吸取5μl进样分析。每个浓度平行3个样本。记录色谱图、QA峰面积As和内标峰面积Ai。计算同一浓度样品的和所有样品的按下列公式计算提取回收率:QA的提取回收率×100% ×(840/750),内标 SLS的提取回收率 RECi=(Ai)B/×100% ×(840/750)。QA低、中、高 3个浓度血浆质控样本的平均提取回收率分别为77.75%、80.90%和80.76%,RSD分别为1.70%、1.78%和4.15%,内标平均提取回收率为104.57%,RSD分别为5.47%。结果表明,该方法中QA和内标的提取回收率精密且可重现。
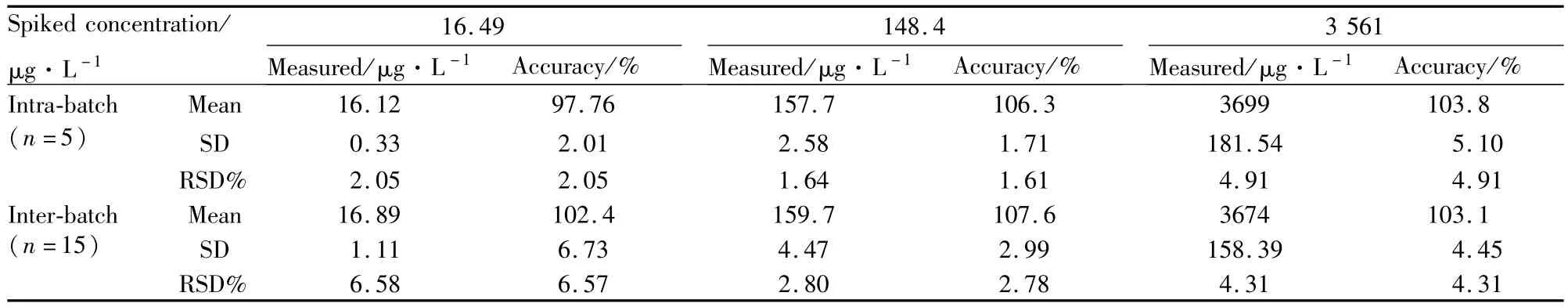
Tab 1 Intra-and inter-batch precision and accuracy for determ ination of QA in human p lasma
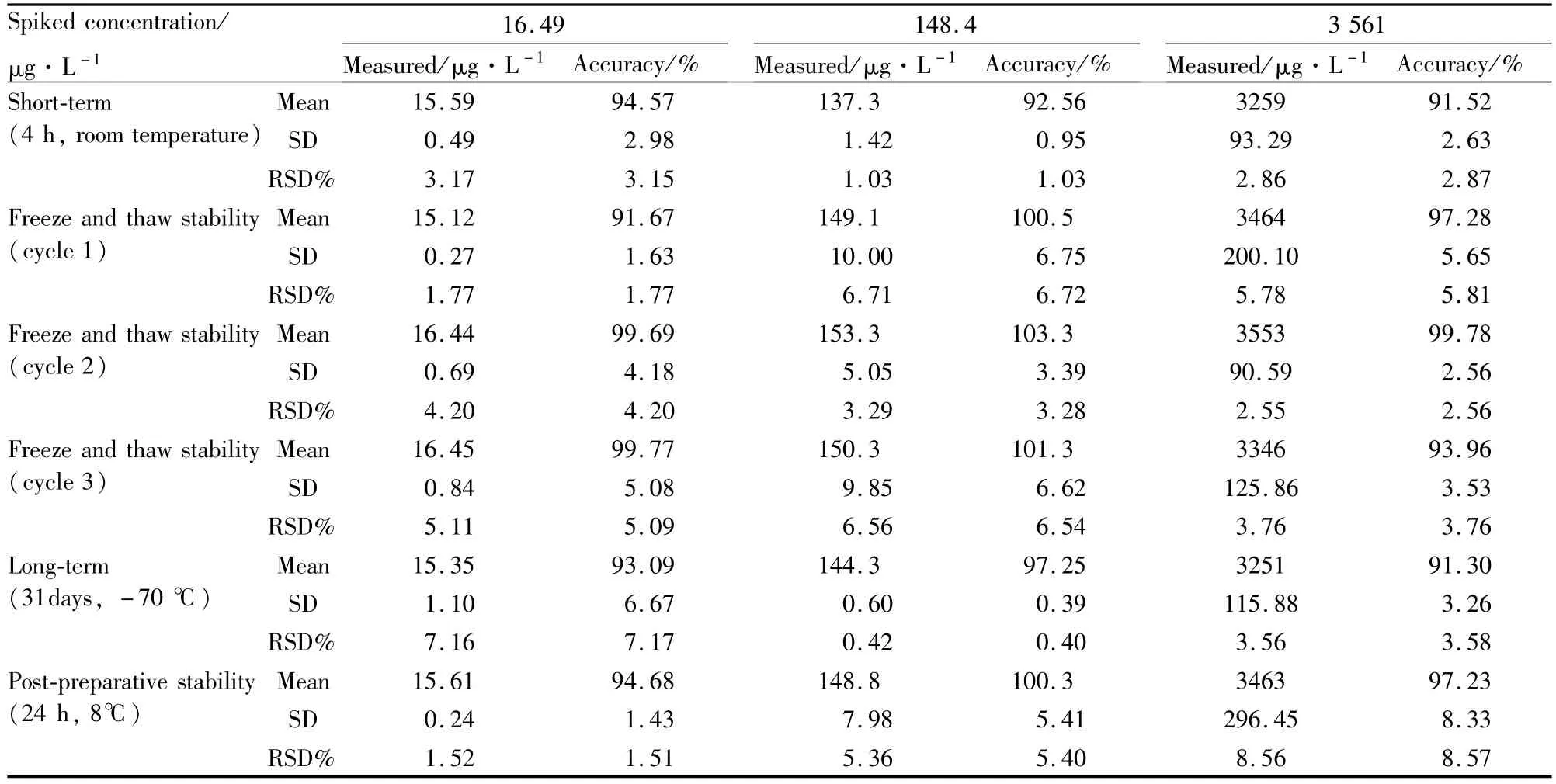
Tab 2 Short-term,freeze-thaw,long-term and post-preparative stability of QA in human p lasma(n=3)
2.4.6 稳定性考察 按“2.3.3血浆质控样本的配制和处理”项配制QA低、中、高3种浓度的血浆质控样本,分别考察血浆样本室温放置4 h、-70℃冰箱中保存并反复冻融3次、-70℃冰箱保存1个月及样品处理后自动进样器(8℃)中放置24 h的稳定性。考察结果详见Tab 2,结果表明,在上述各种条件下样品的稳定性良好。
2.5 应用 36名健康男性受试者,经体检合格,签署知情同意书,并经南京中医药大学附属医院伦理委员会审批同意,均分为低、中、高3个剂量组进行药代动力学实验。3组分别单次静滴剂量为25、50和75 mg的注射用QA后,于静滴前、静滴开始后20、40 min和拔针后 0、10、30、45 min、1、1.5、2、2.5、3、4、5、6、8、12、23 h由肘静脉取血3 ml,置肝素化采血管中,混匀,离心(4 000 r·min-1,4℃,5 min),分离上层血浆,按“2.3.1受试者含药血浆样品处理”项下操作,进样分析测定各血浆样品中QA的浓度。测定结果显示,低、中、高3个剂量组的QA血药浓度范围分别为(10.71~2368)、(11.59~4086)、(28.6~4489)μg·L-1。上述结果表明,所建立的QA血药浓度LC-MS/MS分析测定方法适用于QA人体药代动力学的系统研究。
3 讨论
3.1 分析方法的优化 在方法学建立过程中,我们对色谱柱、流动相组成、样品前处理和内标选择进行了系统全面的优化。结果表明:①Agilent ZORBAX SB C18色谱柱分离效果和峰形优于Thermo syncronis C8柱;② 流动相中加入浓度为0.2 mmol·L-1乙酸铵可提高QA的色谱峰响应强度,而在样品处理过程中,加入0.2 mol·L-1乙酸铵则可以使QA和内标的柱保留时间保持稳定;③ 采用固相萃取(SPE)法处理血浆时,QA的提取回收率明显高于液液萃取法和有机溶剂沉淀法,waters Oasis@HLB小柱提取效果优于Thermo Soly小柱;④QA血浆样品分析过程中易出现色谱峰前延现象,在流动相中和SPE处理上样前的样品中同时加入乙酸铵可改善峰前延问题,但浓度增高会降低QA和内标的质谱响应,故最终优化确定其浓度为0.2 mmol·L-1和0.2 mol·L-1;⑤ 作为定量用内标,SLS和葛根素在本研究的分析条件下色谱峰峰形良好,而芦丁色谱峰有拖尾现象出现;多次反复进样发现,SLS的响应强度稳定,而葛根素的响应值有增大趋势,因此最终选择SLS作为本方法的内标。
3.2 分析方法的改进 与先前报道的QA血浆浓度测定的 LC-MS/MS方法[7]相比,本方法的改进之处在于:① 通过对高浓度QA血样进行稀释比考察,扩大了分析方法可以准确定量的浓度范围。本方法的最终可准确定量范围为8.570~26 150μg·L-1,远高于先前报道的1 000倍浓度跨度。②得益于方法学建立过程的系统优化,检测的灵敏度有所提高,从而减少了血浆样品用量,即从先前报道的1 ml减少至400μl。③ 简化了样品SPE处理过程,洗脱液即可直接进样分析,无需进行吹干、复溶等繁杂操作。上述改进使得本方法能够对低、中、高不同剂量组受试者的QA血药浓度实现简单快速、灵敏准确的测定,从而保证系统的QA人体药代动力学研究的顺利开展。
参考文献:
[1] 李欣志,刘建勋,尚晓泓,等.羟基红花黄色素A对犬急性心肌缺血的保护作用 [J].中国药理学通报,2006,22(05):533-7.
[1] Li X Z,Liu JX,Shang X H,et al.Protective effect of Hydroxysafflor Yellow A on acutemyocardial ischemia in dogs[J].Chin Pharmacol Bull,2006,22(05):533-7.
[2] 夏玉叶,闵 旸,盛雨辰.羟基红花黄色素A对大鼠血栓形成和血小板聚集功能的影响 [J].中国药理学通报,2005,21(11):1400-1.
[2] Xia Y Y,Min Y,Sheng Y C.The effect of Hydroxysafflor Yellow A on the formation of thrombus and platelet aggregation in rats[J].Chin Pharmacol Bull,2005,21(11):1400-1.
[3] Song L,Zhu Y,Jin M,et al.Hydroxysafflor yellow a inhibits lipopolysaccharide-induced inflammatory signal transduction in human alveolar epithelial A549 cells[J].Fitoterapia,2013,84:107-14.
[4] Liu Y,Lian Z,Zhu H,et al.A systematic,integrated study on the neuroprotective effects of hydroxysafflor yellow a revealed by(1)H NMR-based metabonomics and the NF-kappaB pathway[J].Evidence Based Complement Alternat Med:eCAM,2013,2013:1-14.
[5] 杨 静,杨志福,田 云,等.HPLC-UV法测定人血浆中羟基红花黄色素A含量 [J].解放军药学学报,2009,25(1):68-70.
[5] Yang J,Yang Z F,Tian Y,et al.Determination of hydroxysafflor yellow A in human plasma by HPLC-UV[J].Pharm JChin People′s Liberat Army,2009,25(1):68-70.
[6] 曾志平,王实强.高效液相色谱法测定人血浆中羟基红花黄色素A的浓度 [J].中南药学,2010,(1):33-6.
[6] Zeng Z P,Wang SQ.Determination of hydroxysafflor yellow A in human plasma by high performance liquid chromatography[J].Central South Pharm,2010,(1):33-6.
[7] Wen A,Yang J,Jia Y,et al.A rapid and sensitive liquid chromatography-tandem mass spectrometry(LC-MS/MS)method for the determination of hydroxysafflor yellow A in human plasma:application to a pharmacokinetic study[J].JChromatogr B,Analyt Technol Biomed Life Sci,2008,876(1):41-6.
猜你喜欢
中国氯碱(2022年10期)2022-11-22
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
中国现代医药杂志(2020年10期)2020-12-14
化学工程师(2020年7期)2020-09-07
临床检验杂志(电子版)(2020年1期)2020-04-03
发明与创新·中学生(2019年9期)2019-09-12
天津医科大学学报(2019年3期)2019-08-13
中成药(2018年6期)2018-07-11
制造技术与机床(2017年9期)2017-11-27
