
以聚乙二醇/甲醇为二元致孔剂的新型磺酸甜菜碱型两性离子亲水毛细管整体柱的合成与色谱评价
2014-05-08 11:14况媛媛
色谱 2014年4期
况媛媛, 王 彦, 谷 雪, 李 静, 阎 超
(上海交通大学药学院,上海200240)
近年来,随着极性化合物的分离分析在药物分析、药物代谢、食品和环境科学[1-4]等领域越来越受到重视,亲水作用色谱(hydrophilic interaction liquid chromatography,HILIC)得到了快速发展。亲水作用色谱的概念最早由 Alpert[5]于 1990年提出。HILIC可以较好地分析反相色谱不能分离的极性化合物,并且由于它的流动相由有机试剂与水混合而成,弥补了正相色谱对于水溶性物质溶解差、保留时间对流动相中水含量敏感以及与质谱检测器不兼容等缺点[6]。
整体柱(monolithic column)是将单体、交联剂、致孔剂和引发剂等混合通过原位聚合制备而成的棒状连续床层。因其传质速度快、反压低、柱效高而被称为第四代分离介质,已在蛋白质、多肽、核酸等[7]样品的分离分析中显示优势。亲水整体柱集合了亲水作用色谱与整体柱的特点,与反相色谱整体柱相比更适合分离极性物质,为微分离技术提供了更多的选择空间。Lämmerhofer等[8]最早采用 2-甲基丙烯酸二甲胺乙酯(DMAEMA)、2-甲基丙烯酸羟乙酯(HEMA)和亚乙基二甲基丙烯酸酯(EDMA)共聚获得DMAEMA-co-HEMA-co-EDMA亲水整体柱并应用于布洛芬类非甾体抗炎药物的分离。Wang等[9]采用HEMA与季戊四醇三丙烯酸酯(PETA)制备了聚(PETA-co-HEMA)中性整体柱,所制得整体柱表面不带电荷,适合于分离碱性物质[10]。最近,Lin等[11]采用无机单体多面体低聚倍半硅氧烷(POSS-MA)与HEMA合成了有机-无机硅胶杂化亲水整体柱,并成功应用于5种脑啡肽的分离。
以磺酸盐甜菜碱作为聚合物的制备单体[12],因其结构中同时含有季铵基和磺酸基两个活性基团,且二者数目相等,净电荷为零,故不易产生自发聚合,易进行自由基引发聚合。由于其具有良好的化学及热稳定性及pH值耐受范围宽等特点而逐渐引起人们的关注。N,N-二甲基-N-甲基丙烯酰胺基丙基-N,N-二甲基-N-丙烷磺酸内盐(SPP)作为磺酸甜菜碱类两性离子化合物具有很强的亲水性,可用于制备亲水性固定相。
李新燕等[13]以甲基丙烯酸丁酯(BMA)和磺酸甜菜碱类化合物3-(N,N-二甲基-(2-(2-甲基丙-2-烯酰氧基)乙基)铵)丙烷-1-磺酸内盐(SPP)为单体,制备了新型的亲水作用毛细管整体柱,以加压毛细管电色谱为技术平台,无需添加离子对试剂即可用于奶制品中三聚氰胺的定量测定。高也等[14]以SPP与季戊四醇三丙烯酸酯(PETA)为单体,分别用甲醇/1,4-丁二醇和乙醇/乙二醇为致孔剂制备了亲水整体柱,其中用乙醇/乙二醇制备的亲水柱与甲醇/1,4-丁二醇制备的亲水柱相比,渗透性较差,但柱效较高。本文在此基础上,以SPP为单体,PETA为交联剂,偶氮二异丁腈(AIBN)为引发剂,采用聚乙二醇(PEG)/甲醇作为致孔体系制备了一种新型亲水性整体柱,并以毛细管液相色谱(capillary liquid chromatography,cLC)和加压毛细管电色谱(pressurized capillary electrochromatography,pCEC)为平台,应用于核苷类、酚类、胺类等极性化合物的分离,获得了很好的效果。
1 实验部分
1.1 试剂、材料和仪器
SPP和PETA(Sigma-Aldrich,USA);AIBN 和氢氧化钠(NaOH)(中国医药集团上海化学试剂有限公司);PEG(国药集团化学试剂有限公司);甲醇和乙腈(色谱纯,Tedia公司);去离子水(南京娃哈哈饮料有限公司);γ-(甲基丙烯酰氧)(γ-MAPS;Alfa Aesar,USA);熔融石英毛细管(100 μm i.d.×360 μm o.d.,河北永年锐沣色谱器件有限公司)。胸腺嘧啶、尿嘧啶、腺嘌呤、尿苷、肌苷、胞嘧啶、鸟嘌呤、鸟苷(北京百灵威科技有限公司);苯酚、邻苯二酚、间苯二酚、对苯二酚、焦棓酸、间苯三酚、甲酰胺、对甲苯胺、丙烯酰胺、烟酰胺、N,N'-亚甲基双丙烯酰胺(中国医药集团上海化学试剂有限公司)。
TriSep-2100GV加压毛细管电色谱仪(Unimicro Technologies,Inc.,USA),包括柱上紫外检测器、微流控系统、溶剂输送系统、高压电源和数据采集系统;场发射扫描电镜(FEI;Hillsboro,USA);Pore-Master33 Mercury Porosimetry Analyzer全自动压汞仪(Quantachrome,USA);KQ2200B型超声波清洗器(昆山超声仪器有限公司)。
1.2 毛细管整体柱的制备
毛细管的预处理:用配制的0.1 mol/L NaOH溶液冲洗毛细管30 min,再用去离子水冲洗10 min至流出的液体为中性;将冲洗过的毛细管用氮气吹4 h至毛细管完全干燥。将配制好的体积分数为50%的γ-MAPS甲醇溶液充满毛细管,两端用橡胶塞封口,放入25℃恒温水浴中反应12 h,反应完全后取出毛细管,用甲醇冲洗1 h后通氮气吹干,备用。
整体柱的制备:称取适量的PEG与甲醇超声至完全澄清透明,加入适量的 SPP和 PETA,涡旋3 min,置于水浴中超声15 min,加入适量的AIBN,超声10 min,通氮气排气5 min;将聚合液用注射器注入到预处理好的毛细管中,两端用橡胶塞封口,放入60℃水浴中反应20 h。反应结束后将毛细管柱接到高压泵上,用甲醇冲洗1 h以除去未反应的单体与致孔剂。
在柱填料的末端做标记,利用加热的电阻丝在标记处烧一段2~3 mm的窗口,截取2 cm的毛细管柱做场发射扫描电镜(scanning electron microscope,SEM)实验。
1.3 整体柱性能的考察
1.3.1 渗透率K的测定
所有试验均在室温下进行。在毛细管液相色谱模式下,根据公式× η[15]计算甲醇通过整体柱时的渗透率K,其中μ为甲醇通过整体柱的线性流速,η为甲醇在室温下的动态黏滞度(0.58 mPa)[16],L为整体柱的有效长度,ΔP为甲醇通过整体柱的柱压。
1.3.2 整体柱色谱性能的考察
通过毛细管液相色谱和加压毛细管电色谱分离不同极性化合物考察毛细管整体柱的色谱性能。毛细管整体柱规格为250 mm(总长500 mm)×100 μm,分流管规格为2 m×50 μm,分流前进样环体积约为1 μL,分流比约为200∶1,实际进样量为5 nL。
2 结果与讨论
2.1 亲水整体柱聚合液组成比例的优化
致孔剂的种类和用量在很大程度上影响整体柱的孔径大小及分布,从而影响整体柱的渗透性。致孔剂的选择原则一般为致孔剂要与单体互溶,且不与单体反应;反应完全后易于去除等。PEG是一种无毒高分子聚合物,具有良好的水溶性,并与多种有机组分有极好的互溶性,一般作为模板剂应用于硅胶整体柱的溶胶-凝胶制备过程,而较少用于聚合物整体柱的制备过程[17]。但由于它的相对分子质量范围可以从几百到几万,可以更好地调节聚合物的孔尺寸、密度和体积,因此在本研究中选择PEG/甲醇为致孔体系。
单体与致孔体系比例的优化。依次按照单体与致孔体系的质量比为 1∶4、1∶3、1∶2.5、1∶2 制备 A、B、C、D 4批整体柱(见表1),制备过程中其他反应物含量均保持不变。结果表明,随着致孔体系含量的减少,整体柱的通透性呈下降趋势,柱效呈上升趋势。其中,当单体与致孔体系的质量比为1∶2时,聚合液呈现不溶解的状态,无法制备均匀连续的整体柱。综合考虑柱效和柱的通透性,在后续的实验中选择单体与致孔体系的质量比为1∶2.5。
致孔体系内部比例的优化。依次按照PEG-6000 与甲醇的质量比为 1∶3、1∶2.5、1∶2、1∶1.5 制备E、F、C、G 4批整体柱(见表1)。其中随着PEG含量的增加,渗透性与柱效呈现升高趋势。当PEG与甲醇的质量比为1∶1.5时,聚合液不溶解,达到饱和状态,无法制备拥有均一柱床的整体柱。综合考虑柱效与渗透性二者的最优化,在后续的实验中选择PEG与甲醇的质量比为1∶2。
交联剂与单体比例的优化。按照交联剂与单体的质量比分别为 1∶2、1∶1.5、1∶1、1∶0.5 制备 H、I、C、J 4批整体柱(见表1)。由表1可见,随着交联剂比例的增大,整体柱的渗透性越来越小,柱效增加;当PETA与SPP的质量比为1∶0.5时,交联剂比例过大导致整体柱渗透性太差,甲醇无法将整体柱冲开。综上所述,在后续的实验中选择单体与交联剂的质量比为1∶1。
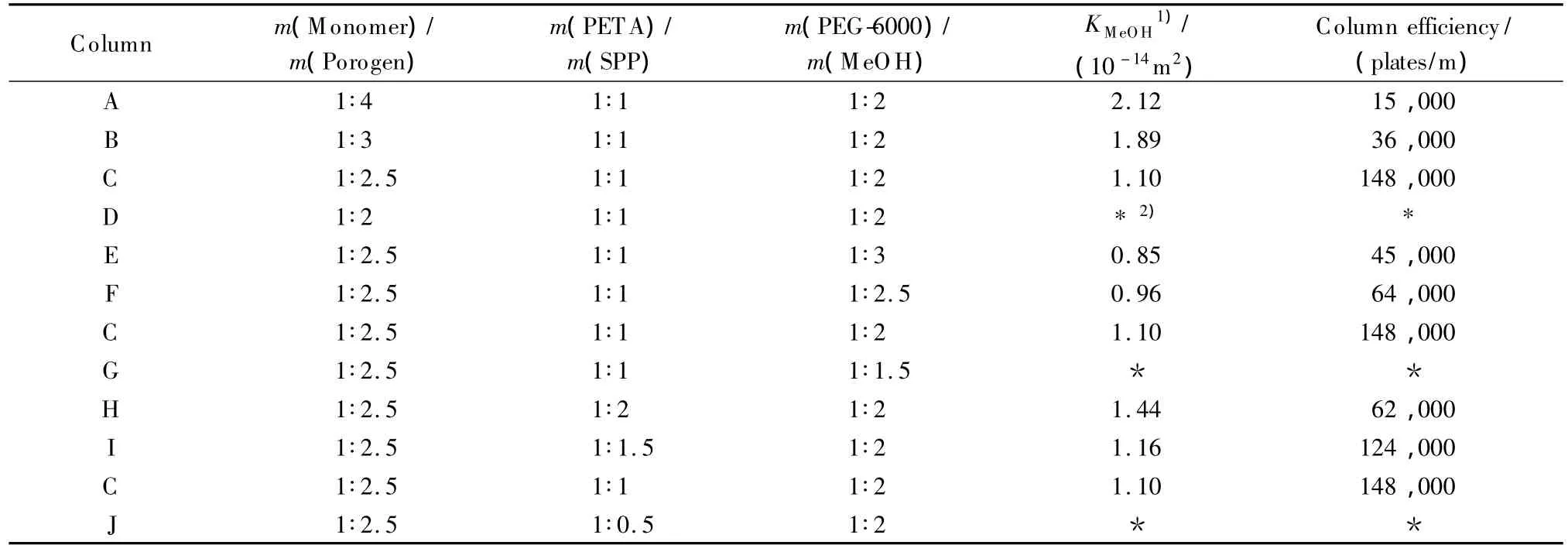
表1 制备亲水整体柱聚合液的组成Table 1 Composition of polymerization mixtures for the preparation of hydrophilic monolithic columns
PEG随着聚合度的增加,熔点和黏度也都相应增大,我们选取了相对分子质量从2 000到10 000的PEG作为致孔剂制备亲水整体柱,考察PEG的相对分子质量对整体柱的孔径、柱效以及渗透性等因素的影响(见表2)。可见,由不同相对分子质量的PEG制备而成的整体柱存在差异,随着PEG相对分子质量的增加,整体柱的渗透性与柱效均呈升高趋势,这可能是因为随着相对分子质量的增大,致孔剂溶液中存在着更大的空间位阻,导致贯穿孔增多,渗透性增大,但用PEG-6000制备的柱C的柱效高于用PEG-10000制备的柱M,柱效达到148 000塔板/m。综合以上所有因素,选取亲水整体柱的最优化制备条件为:单体与致孔剂的质量比为1∶2.5,致孔剂中PEG-6000与甲醇的质量比为1∶2,SPP与 PETA的质量比为1∶1,AIBN为总质量的0.1%。

表2 以不同相对分子质量的PEG制备的整体柱的渗透性及柱效Table 2 Permeabilities(K)and column efficiencies of the monolithic columns prepared using PEG with different relative molecular masses
2.2 整体柱的表征
2.2.1 整体柱的形貌表征
用场发射扫描电镜表征最终制得的聚合物亲水整体柱的骨架与大孔尺寸。从图1可以看出,在优化条件下制备的整体柱拥有均一的孔结构,固定相与毛细管管壁牢固地结合成一个整体,没有发生脱落的现象;整体结构中有微米级的贯穿孔,保证了整体柱有良好的通透性。
2.2.2 整体柱的孔径分布
通过压汞法测得整体柱的平均孔径为208 nm左右。根据文献[18]可以计算出整体柱的孔隙率为0.72。
由以上数据可以看出,整体柱具有较大的孔径及较高的孔隙率,通透性相对较好。
2.3 亲水整体柱的色谱性能
在cLC模式下,在不同流速下对制备的整体柱C拟合范第姆特方程曲线(见图2)。当线性流速达到0.778 mm/s时,最高柱效为1.02 ×105塔板/m。另外,在cLC模式下对整体柱C的亲水性进行了考察。当流动相中乙腈含量为95%时,甲苯、丙烯酰胺和硫脲3种物质的出峰顺序与极性相关,极性越大出峰越晚;在反相色谱模式下不保留的硫脲在17 min左右出峰,说明整体柱C具有良好的亲水性。

图1 亲水整体柱C场发射扫描电镜照片Fig.1 SEM photographs of the hydrophilic monolithic column C
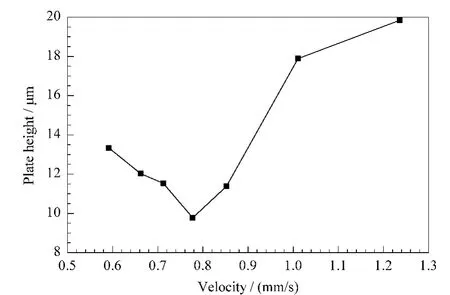
图2 亲水整体柱C的范第姆特方程曲线Fig.2 van Deemter plot of the height equivalent to a theoretical plate height as a function of flow rate of hydrophilic monolithic column CConditions:cLC mode;column dimension,250 mm(total length 500 mm)×100 μm;mobile phase,ACN/H2O(88/12,v/v)containing 10 mmol/L ammonium formate;detection,UV at 214 nm.
2.4 亲水整体柱的重复性与稳定性
选取甲苯与丙烯酰胺为样品,在cLC模式下考察亲水整体柱的保留因子与柱效的重复性与稳定性,结果如表3所示。其中整体柱的日内与日间RSD均小于5%。同时考察不同批次间亲水整体柱保留因子与柱效的重复性,6根整体柱的RSD均在可接受范围内,表明所制备的整体柱具有良好的重复性与稳定性。
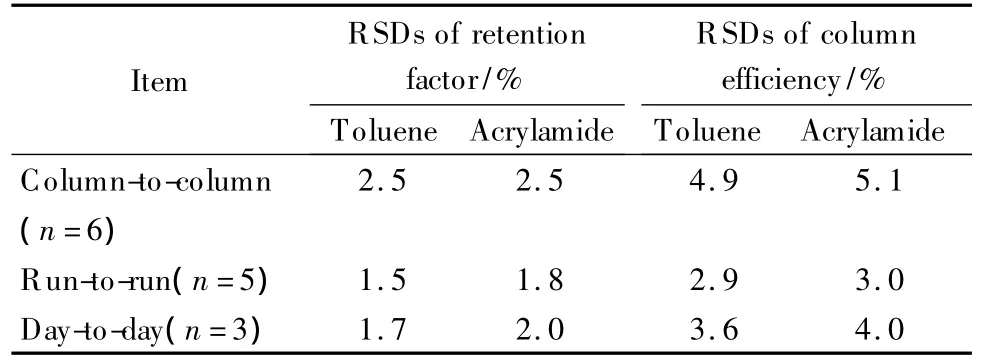
表3 亲水整体柱C的保留因子与柱效的重复性Table 3 R eproducibilities of retention factor and column efficiencies on column C
2.5 对于分离机理的探讨
HILIC分离机理[19]被认为是多种机理的混合作用,如氢键、吸附、偶极矩为初级作用机理;而带电荷的固定相可以提供第二个十分重要的有选择性的保留作用,也就是待分离物质与固定相上的电荷之间的静电作用。并且HILIC的作用机理受到许多因素的影响,最主要的是受到固定相的种类、流动相中有机相的含量、流动相的pH以及缓冲盐离子浓度等因素的影响。
为了研究磺酸甜菜碱两性离子亲水整体柱的分离机理,我们选取甲苯、丙烯酰胺、硫脲3种极性相差较大的物质,通过改变流动相中有机相乙腈的含量来考察3种物质保留时间的变化趋势。如图3所示,在流动相中乙腈含量超过39%时,硫脲的保留时间比甲苯和丙烯酰胺长;且在乙腈含量由95%降至39%的过程中,硫脲的保留时间也在逐渐降低;在乙腈含量继续降至20%的过程中,硫脲的保留时间下降趋势减缓,趋于稳定状态。甲苯因其非极性较强,在乙腈含量从95%降到50%的过程中,它的保留时间先略微增长然后基本保持不变;当乙腈含量低于50%后,随着乙腈含量的降低,甲苯的保留时间急剧升高;当乙腈含量低于39%时,甲苯在硫脲后面出峰。丙烯酰胺由于其极性介于甲苯与硫脲之间,所以随着流动相中乙腈含量的升高呈现大致升高的趋势,在乙腈含量从50%上升到95%的过程中,其保留时间超过甲苯。结果表明,当乙腈含量大于50%时,整体柱的分离机理表现为亲水作用机理[20]。
2.6 亲水整体柱的应用
2.6.1 核苷类化合物的分离
由于核苷类化合物具有强极性,因而很难在反相色谱模式下得到较好的分离。为此,我们采用亲水模式,分别从流动相pH、盐浓度以及流动相中乙腈含量等分离条件对核苷类物质的分离进行了优化。
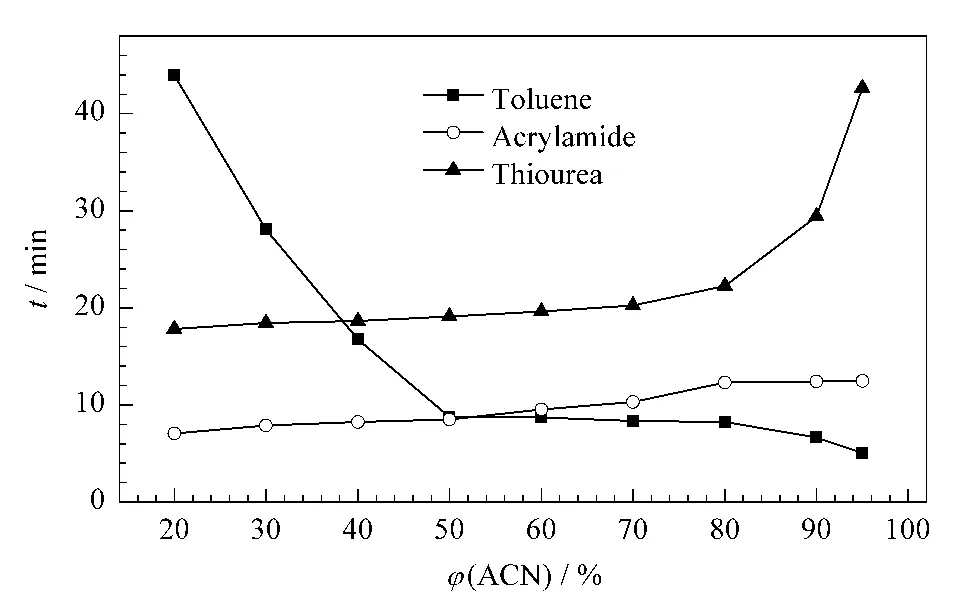
图3 流动相中乙腈含量对甲苯、丙烯酰胺、硫脲保留时间的影响Fig.3 Influence of the ACN content in mobile phase on the retention times of toluene,acrylamide and thioureaConditions:cLC mode;column dimension,250 mm(total length 500 mm)×100 μm;mobile phase,ACN/H2O system with different volume ratios,containing 10 mmol/L ammonium formate;flow rate,0.05 mL/min;detection,UV at 214 nm.
在cLC模式下8种核苷类化合物随pH的变化趋势大致相同,依次调整流动相的pH 为3、5、7、9,8种物质的保留因子都是先增大后减小,当pH值为7时达到最大值。各样品的pKa值分别为胸腺嘧啶9.94,尿嘧啶 9.5,腺嘌呤 9.8,尿苷 9.25,肌苷 8.9,胞嘧啶12.16,鸟嘌呤9.92,鸟苷9.36。考虑到当流动相的pH为3、5、7时,亲水保留机理和离子交换保留机理共同作用,此时样品均呈正电性,更易与磺酸根发生离子交换,但磺酸根随着pH增大会呈解离状态,而核苷类物质在此时也都是正电性,所以离子交换程度增大;pH在7~9范围时核苷类物质的正电性减弱,离子交换程度降低,因此保留减弱。
同样,我们也考察了流动相中甲酸铵的浓度对核苷类物质保留的影响。实验中将流动相的甲酸铵浓度从10 mmol/L一直升高到70 mmol/L,这些核苷类物质在整体柱上的保留先增大后减小,当甲酸铵浓度为40 mmol/L时保留因子最大;当甲酸铵浓度从10 mmol/L升高到40 mmol/L时,样品的保留逐渐增大。根据Alpert[21]的理论,在流动相的盐浓度逐渐增大的情况下,越来越多的盐离子进入固定相的水化层,从而增大了固定相的容积,使样品保留增加。当甲酸铵浓度从40 mmol/L增加到70 mmol/L时,水化层中盐离子数量足够多,以至于与固定相中的磺酸根离子发生离子交换,使样品与固定相的离子交换减弱,保留也随之减弱。
通过以上考察我们选取pH为7,流动相中甲酸铵浓度为40 mmol/L作为核苷类物质的分离条件。另外,我们也考察了不同含量的乙腈对分离的影响。如图4所示,在95%乙腈条件下,各物质因结构相似很难达到有效分离;在90%乙腈条件下,峰形与分离度都有所改善,其中只有尿苷与肌苷未达到基线分离;当乙腈浓度减少为85%时,8种物质的保留时间较为接近,分离度变差。所以我们选择90%乙腈作为后续的实验条件。
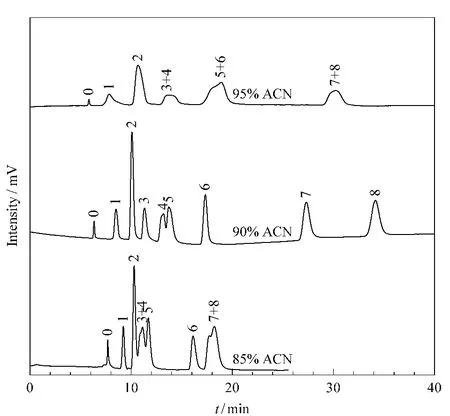
图4 乙腈含量对核苷化合物在亲水整体柱C上分离的影响Fig.4 Effect of ACN content on the retention of nucleosides on hydrophilic monolithic column CConditions:cLC mode;column dimension,250 mm(total length 500 mm)×100 μm;mobile phase,ACN/H2O system with different volume ratios,containing 40 mmol/L ammonium formate;flow rate,0.05 mL/min;detection,UV at 254 nm.Peaks:0.toluene;1.thymine;2.uracil;3.ademine;4.uridine;5.inosine;6.cytosine;7.guanine;8.guanosine.
在加压毛细管电色谱模式下,整体柱两端施加正电。如图5所示,随着电压的增大,峰形变好,分离度提高,柱效也有所增加,其中电压为10 kV时8种核苷类化合物都得到基线分离;15 kV时分离度并未继续提高,而峰形却不如10 kV时。综上所述,8种核苷化合物的最优分离条件为pH为7,甲酸铵浓度为40 mmol/L,电压为10 kV。
2.6.2 其他极性化合物的分离
酚类化合物的分离 图6为亲水整体柱C对酚类化合物的分离图。结果显示,随着化合物中羟基数目的增加,保留时间相应增加,6种酚类混合物得到基线分离,出峰顺序也与文献[22]报道一致。
胺类化合物的分离 在毛细管液相色谱模式下对胺类极性小分子化合物进行了分离并获得了理想效果。如图7a所示,其中分离N,N'-亚甲基双丙烯酰胺的柱效可达到2.4×105塔板/m,而图7b中,乙酰苯胺与N,N'-亚甲基双丙烯酰胺的分离度为1.4,没有达到基线分离。
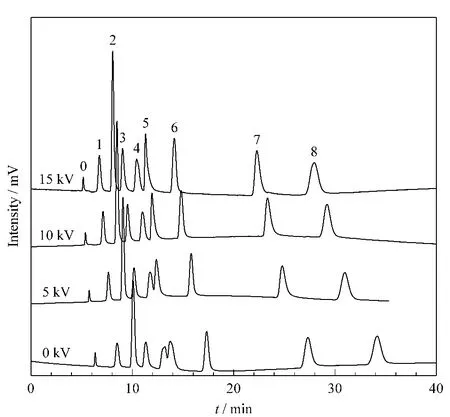
图5 分离电压对核苷化合物在亲水色谱柱C上分离的影响Fig.5 Effect of separation voltage on the retention of nucleosides on hydrophilic monolithic column CConditions:pCEC mode;mobile phase,ACN/H2O(90/10,v/v)containing 40 mmol/L ammonium formate,pH 7;others are the same as in Fig.4.Peak identifications are the same as in Fig.4.
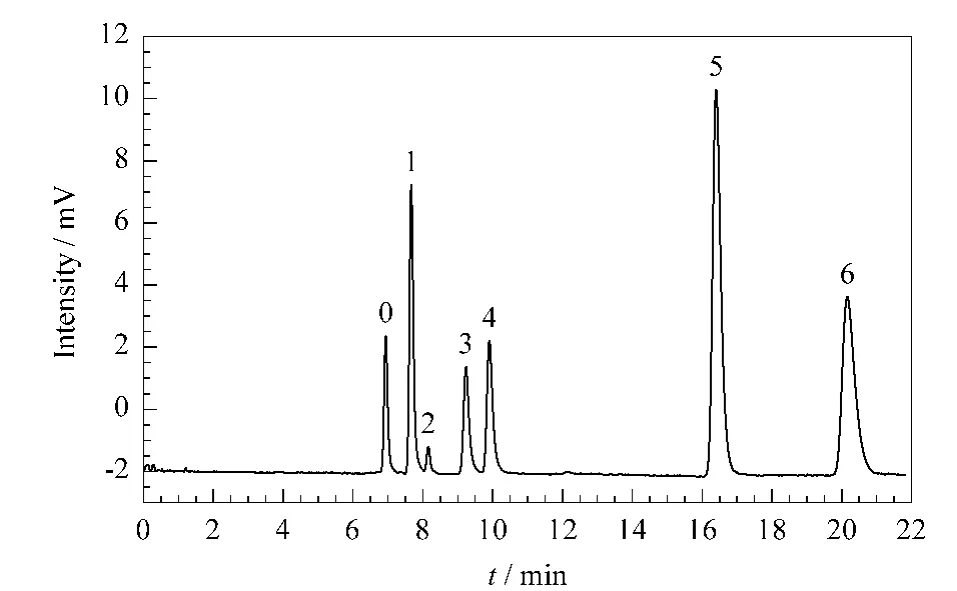
图6 亲水色谱柱C对酚类化合物的分离Fig.6 Chromatogram of phenols on hydrophilic monolithic column CConditions:cLC mode;mobile phase,ACN/H2O(95/5,v/v)containing 10 mmol/L ammonium formate;detection,UV at 214 nm;others,same as in Fig.4.Peak identifications:0.toluene;1.phenol;2.quinol;3.catechol;4.resorcinol;5.pyrogallol;6.phloroglucinol.
杂环类化合物的分离 杂环类化合物具有一定的生物活性,在医药领域中有广泛的应用。由于杂环类化合物结构较为相近,分离困难,我们用制备的新型亲水整体柱C在加压电色谱模式下在7 min内分离了4种杂环类化合物,如图8所示,4种物质达到了基线分离。
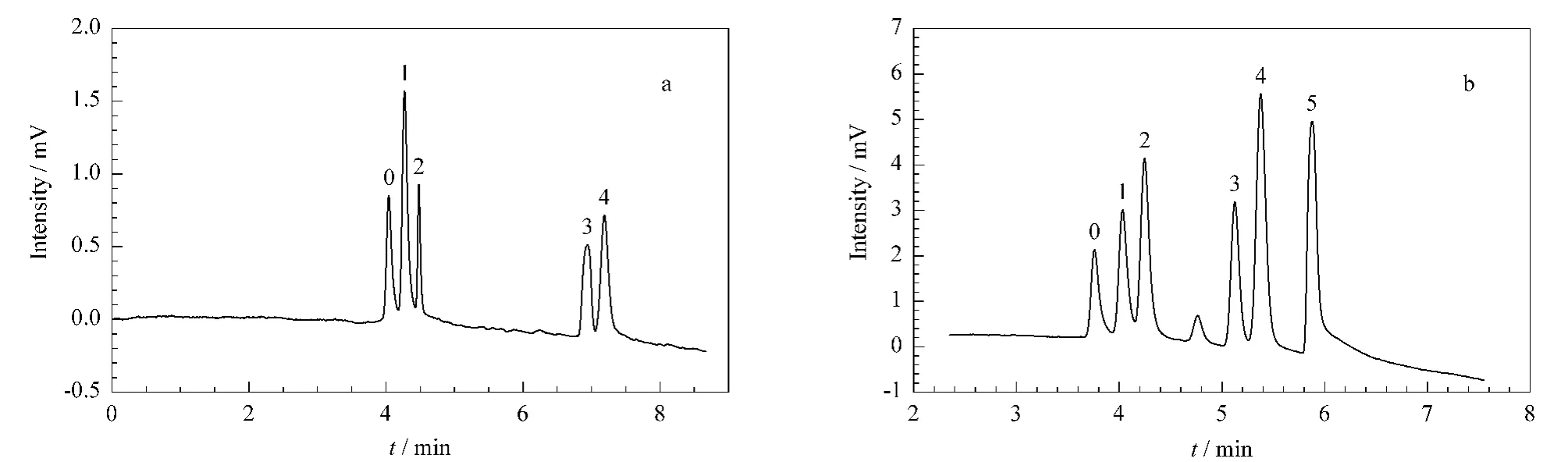
图7 亲水色谱柱C对胺类化合物的分离Fig.7 Chromatograms of amides on hydrophilic monolithic column CThe conditions are the same as in Fig.6.Peak identifications:a.0.toluene;1.paratoluidine;2.N,N'-dimethylenebisacrylamide;3.formamide;4.thymine.b.0.toluene;1.acetanilide;2.N,N'-dimethylenebisacrylamide;3.acrylamide;4.nicotinamide;5.formamide.
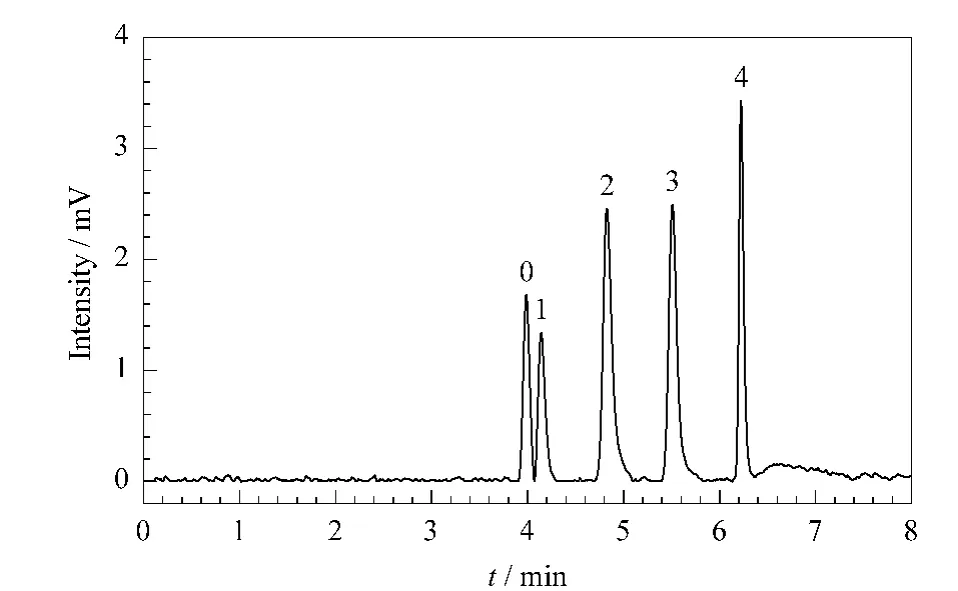
图8 亲水色谱柱C对杂环类化合物的分离Fig.8 Electrochromatogram of heterocyclics on hydrophilic monolithic column CConditions:pCEC mode;mobile phase,ACN/H2O(88/12,v/v)containing 10 mmol/L ammonium formate;backpressure,6.4 MPa;applied voltage,20 kV;others,same as in Fig.7.Peak identifications:0.toluene;1.caffeine;2.theophyline;3.nicotin;4.thymine.
3 结论
本文以PEG/甲醇为二元致孔剂,SPP与PETA为单体,原位聚合制备了亲水整体柱;所制备的整体柱机械强度好,孔径分布均匀,渗透性好,反压低,并且具有良好的亲水性,能够很好地分离酚类、胺类、杂环类、核苷类等极性较大的物质,可以作为反相色谱应用的补充,也可为亲水-反相色谱联用提供新的思路。
[1] Chen H S,Shen M,Chen L Y.Chromatographia,2011,73(7/8):767
[2] Ikegami T,Tomomatsu K,Takubo H,et al.J Chromatogr A,2008,1184(1/2):474
[3] van Nuijs A L N,Tarcomnicu I,Covaci A.J Chromatogr A,2011,1218(35):5964
[4] Lü H X,An H T,Xie Z H.Int J Biol Macromol,2013,56:89
[5] Alpert A J.J Chromatogr A,1990,499:177
[6] Guo Z M,Zhang X L,Xu Q,et al.Chinese Journal of Chromatography(郭志谋,张秀莉,徐青,等.色谱),2009,27(5):675
[7] Bai L G,Niu W J,Yang G L.Chinese Journal of Chromatography(白立改,牛文敬,杨更亮.色谱),2013,31(4):303
[8] Lämmerhofer M,Svec F,Fréchet J M J.J Chromatogr A,2001,925(1/2):265
[9] Wang X C,Lin X C,Xie Z H,et al.J Chromatogr A,2009,1216(21):4611
[10] Lin X C,Lin J,Wang J B,et al.Chinese Journal of Chromatography(林旭聪,林葭,王家斌,等.色谱),2010,28(3):284
[11] Lin X C,Wang X,Zhao T T,et al.J Chromatogr A,2012,1260:174
[12] Camilla V,Knut I.Macromolecules,2000,33(7):2539
[13] Li X Y,Wang Y,Gu X,et al.Chinese Journal of Chromatography(李新燕,王彦,谷雪,等.色谱),2010,28(3):231
[14] Gao Y,Wang Y,Wang C R,et al.Chinese Journal of Chromatography(高也,王彦,王超然,等.色谱),2011,30(5):487
[15] Bristow P A,Knox J H.Chromatographia,1977,10(6):279
[16] Lide D R.CRC Handbook of Chemistry and Physics.80th ed.S.l.:CRC Press,1999
[17] Ming L C,Li M L,Bi F Y,et al.J Chromatogr A,2012,1230:54
[18] Huang X J,Wang Q Q,Huang B L.Chinese Journal of Analytical Chemistry(黄晓佳,王秋泉,黄本立.分析化学),2005,33(4):467
[19] Andreas E K,Yiannis C F,Constantine D S.J Chromatogr A,2011,1218(20):2871
[20] Lin J,Huang G H,Lin X C,et al.Electrophoresis,2008,29(19):4055
[21] Alpert A J.Anal Chem,2008,80(1):62
[22] Lin J,Liu S F,Lin J,et al.J Chromatogr A,2011,1218(29):4671
猜你喜欢
理化检验-化学分册(2020年12期)2021-01-26
中成药(2017年5期)2017-06-13
中成药(2017年5期)2017-06-13
浙江化工(2015年2期)2015-11-23
体育世界(学术版)(2015年3期)2015-07-01
化工科技(2015年6期)2015-06-09
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
中国当代医药(2015年9期)2015-03-01
肝博士(2015年2期)2015-02-27
热喷涂技术(2014年4期)2014-11-08
