
灯盏花乙素和灯盏花甲素合成中糖苷化反应研究
2014-04-26 08:26张志朋杨兆祥李鹏辉
亚太传统医药 2014年10期
张志朋,杨兆祥,李鹏辉,张 伟
(1.昆明医科大学 药学院,云南 昆明 650500; 2.昆明制药集团药物研究院,云南 昆明 650106)
灯盏花乙素和灯盏花甲素合成中糖苷化反应研究
张志朋1,杨兆祥2,李鹏辉2,张 伟2
(1.昆明医科大学 药学院,云南 昆明 650500; 2.昆明制药集团药物研究院,云南 昆明 650106)
灯盏花素是由灯盏细辛中提取而来的一类总黄酮物质,其主要成分包括灯盏花乙素和灯盏花甲素。灯盏花乙素是灯盏花类药品的主要活性成分,而目前关于灯盏花甲素和灯盏花乙素的合成方法及药理活性的研究报道还较少。糖苷化反应是合成灯盏花乙素和灯盏花甲素过程中的关键步骤,传统糖苷化反应路线较长、成本较高,不利于工业化生产。现对灯盏花乙素和灯盏花甲素合成中的糖苷化反应进行深入研究,优化合成工艺。经实验验证,该糖苷化反应路线可降低成本,简化步骤,利于工业化生产。
灯盏花乙素;灯盏花甲素;糖苷化反应;药物合成
灯盏花即灯盏细辛,又名东菊,是菊科植物短草飞蓬ErigermbreviscaPus (vant.) Hand-Mazz的干燥全草,主要分布于我国西南省区,如云南、四川、贵州、广西、湖南、西藏等。其最早记载于《滇南本草》中,1977年版《中华人民共和国药典》一部也曾予以收载[1]。灯盏花乙素是从灯盏细辛中提取的一类天然总黄酮,其主要成分为灯盏花乙素和灯盏花甲素,以灯盏花乙素为主,含有较少量的灯盏花甲素[2]。灯盏花乙素具有多种药理活性,可用于高血压、血栓、心肌梗塞、脑梗、心律失常、脑缺血等心血管疾病的治疗[3-7]。灯盏花甲素相关文献报道还较少,其在心脑血管疾病方面的疗效不及灯盏花乙素,但可用于老年痴呆、回流性食管炎、溃疡等疾病的治疗[8-9]。目前市售的灯盏花乙素原料药制备主要以植物提取为主,但有提取收率低、提取成本高、环境污染严重的缺点。随着市场需求的扩大,植物提取的天然灯盏花乙素已远不能满足市场需求,因此化学合成灯盏花乙素将有着广阔的市场前景。在灯盏花乙素和灯盏花甲素的合成过程中,糖苷化反应是关键步骤。本文对该步糖苷化反应进行了大量研究,优化了合成工艺,且经过试验验证,优化的合成步骤可用于工业化生产。本文将以实际操作为例,探讨不同受体和不同催化剂对糖苷化反应的影响。
灯盏花乙素和灯盏花甲素的结构式如图1所示。实验中所涉及的化合物的结构式如图2所示。
1 方法与结果
糖苷的合成一般是由糖基给体与不同受体在催化剂的作用下进行的。在糖化学合成中,目前已发展出20多种合成糖苷键的方法[10-12],但任何一个糖苷化方法的适用性都是有限的,难以满足糖苷键的多样化要求。目前最常用的糖基给体为糖基、卤代糖基、糖基三氯乙酰亚胺酯和硫苷。文献报道的糖苷化反应方法有经典的 Koenigs-Knorr 法[10]、相转移催化(PTC)法[11]、糖基三氯乙酰亚胺酯法[12]、三氟化硼乙醚催化法[13]、酶催化法等[14]。
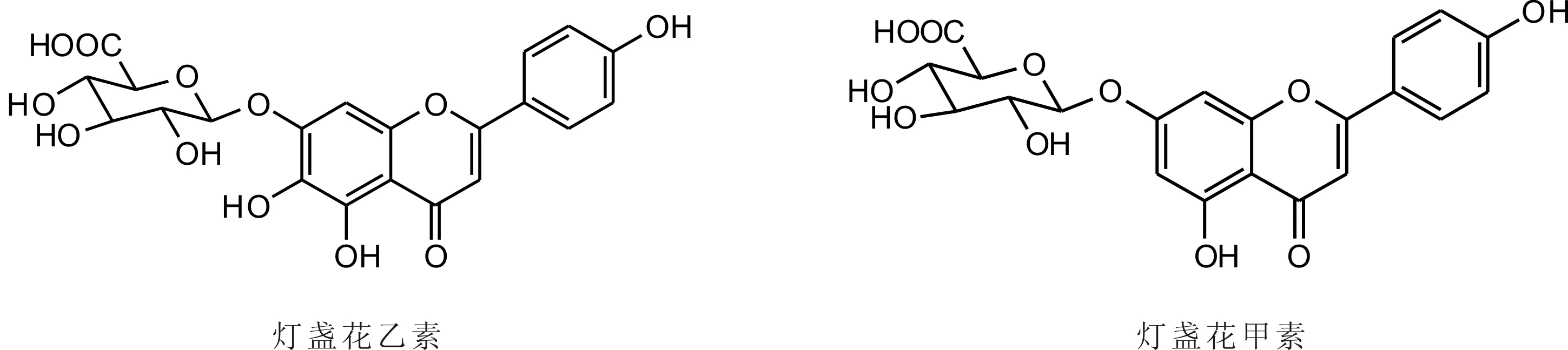
图1 灯盏花乙素和灯盏花甲素结构式
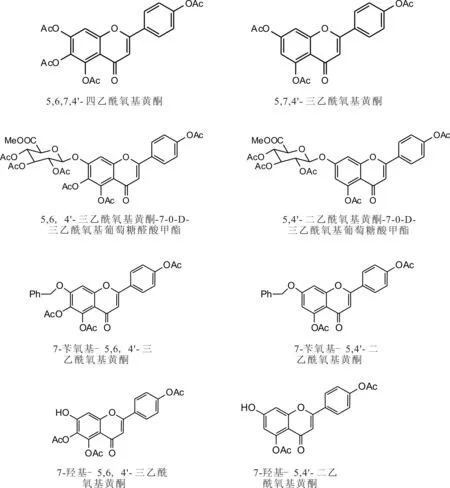
图2 实验中所涉及化合物的结构式
本文通过大量实验探索和工艺改进,采用重金属催化法和相转移催化法,很好地实现了全乙酰化野黄芩素和全乙酰化芹菜素的糖苷化。该糖苷化方法收率可观、重现性好、操作简便、产品纯度高,尤其是使用相转移催化剂进行糖苷化反应可大大降低反应成本,实验验证其可实现工业化生产。本文以贵金属催化法(Koenigs-Knorr 法)和相转移催化法为例,比较了二者运用于不同受体糖苷化反应中的差异。
1.1 不同催化剂对化合物Ⅰ或Ⅱ为原料进行糖苷化反应的影响
1.1.1 重金属盐催化
(1)重金属盐催化剂在灯盏花乙素合成中糖苷化反应的应用[15]。在干燥洁净的250mL圆底烧瓶中依次加入化合物Ⅰ4.54g(0.01moL)、喹啉70mL、无水硫酸钠9g、碳酸银3.6g和溴代糖11.4g,15~30℃下搅拌反应2h后再补加α-溴代三乙酰氧基葡萄糖醛酸甲酯(溴代糖)2.3g,反应6~15h,TLC监测反应是否完全。抽滤,滤饼用二氯甲烷洗至滤液无色,用稀盐酸溶液中和滤液至pH呈中性,加二氯甲烷210mL,水400mL萃取,分离出二氯甲烷层及水层,水层再分别用140mL、70mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液,加水洗涤2~3次。二氯甲烷液用无水硫酸钠干燥,再次抽滤,除去无水硫酸钠及残余的碳酸银,减压蒸干二氯甲烷,得到紫褐色溶液。再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅲ(5,6,4'- 三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)4.91g,收率67.3%,色谱纯度为95%以上。反应方程式如图3所示。
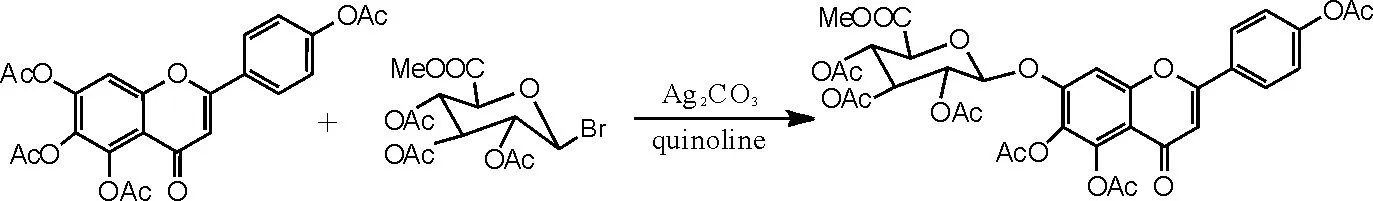
图3 重金属盐催化灯盏花乙素合成反应式
1H-NMR (500MHz, DMSO), δ (ppm): 8.10 (2H, d, J=8.6 Hz), 7.53 (1H, s), 7.37 (2H, d, J= 8.6Hz), 6.88 (1H, s), 5.88 (1H, d, J= 7.8Hz), 5.47 (1H, m), 5.20 (1H, m), 5.10 (1H, m), 4.84 (1H, d, J=10Hz), 3.65 (3H, s), 2.33 (3H, s), 2.31 (3H, s), 2.27 (3H, s), 2.02 (3H, s), 2.01 (3H, s), 1.99(3H, s)。
(2)重金属盐催化剂在灯盏花甲素合成中糖苷化反应的应用[15]。在干燥洁净的250mL圆底烧瓶中加入化合物Ⅱ3.96g(0.01moL),再加入喹啉60mL、无水硫酸钠8g、碳酸银3.2 g和溴代糖8g,15~30℃下搅拌反应2h后再补加α-溴代三乙酰氧基葡萄糖醛酸甲酯(溴代糖)2g,反应6~15h后TLC监测反应是否完全。抽滤,滤饼用二氯甲烷洗至滤液无色,用稀盐酸溶液中和滤液至pH呈中性,加二氯甲烷180mL、水200mL萃取。分离出二氯甲烷层及水层,水层再分别用120mL、60mL二氯甲烷各萃取1次。合并3次萃取的二氯甲烷液,加水洗涤2~3次,二氯甲烷液用无水硫酸钠干燥,再次抽滤,除去无水硫酸钠及残余的碳酸银。减压蒸干二氯甲烷,得到紫褐色溶液,再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)4.23g,收率63.1%,色谱纯度为95%以上。1HNMR(500MHz,DMSO),δ(ppm):8.11(2H,d,J=8.5HZ),7.36(2H,s),7.34(1H,s),6.87(1H,d,J=2.25Hz),6.84(1H,s),5.93(1H,d,J=7.69Hz),5.44(1H,t,J=9.53Hz),5.16(1H,m),5.11(1H,t,J=9.69Hz),4.78(1H,d,J=9.83Hz),3.63(3H,s),2.31(3H,s), 2.30(3H,s), 2.02(3H,s), 2.00(6H,s)。反应方程式如图4所示。
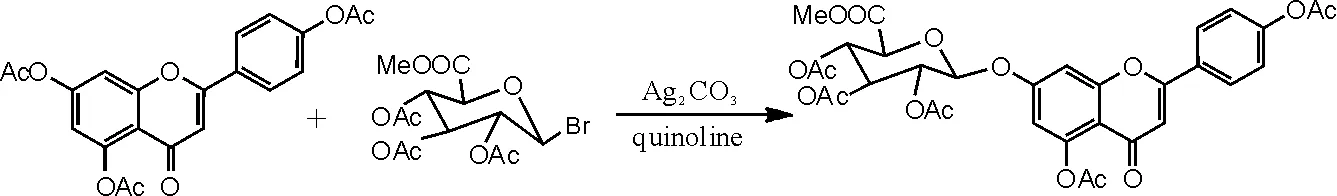
图4 重金属盐催化灯盏花甲素合成反应式
1.1.2 相转移催化剂催化
(1)相转移催化剂在灯盏花乙素合成之糖苷化反应中的应用[16-17]。在干燥洁净的250mL圆底烧瓶中依次加入化合物Ⅰ4.54g(0.01moL)、DMF90mL、碳酸钾9g和相转移催化剂TBAB 3.6g,15~30℃下搅拌10min后加入溴代糖11.4g,反应2h后再补加溴代糖2.3g,反应6~10h后TLC监测反应是否完全。抽滤,滤饼用二氯甲烷洗至滤液无色,加二氯甲烷270mL、水400mL萃取。分离出二氯甲烷层及水层,水层再分别用180mL、90mL二氯甲烷各萃取1次。合并3次萃取的二氯甲烷液,加水洗涤2~3次,二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,得到紫褐色溶液,再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅲ(5,6,4'-三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)4.45g,收率61.1%,色谱纯度为95%以上。1H-NMR (500MHz, DMSO), δ (ppm): 8.10 (2H, d, J=8.6 Hz), 7.53 (1H, s), 7.37 (2H, d, J= 8.6Hz), 6.88 (1H, s), 5.88 (1H, d, J= 7.8Hz), 5.47 (1H, m), 5.20 (1H, m), 5.10 (1H, m), 4.84 (1H, d, J=10Hz), 3.65 (3H, s), 2.33 (3H, s), 2.31 (3H, s), 2.27 (3H, s), 2.02 (3H, s), 2.01 (3H, s), 1.99(3H, s)。反应方程式如图5所示。
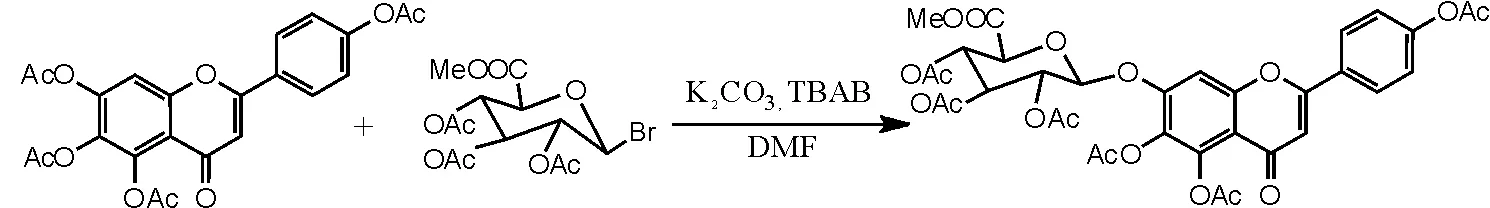
图5 相转移催化剂催化灯盏花乙素合成方程式
(2)相转移催化剂在灯盏花甲素合成中糖苷化反应中的应用[16-17]。在干燥洁净的250mL圆底烧瓶中加入化合物Ⅱ3.96g(0.01moL),再依次加入丙酮120mL、碳酸钾10g,50℃下回流1~2h,待温度降至30℃左右,加入相转移催化剂TBAB 3.2g,搅拌10min左右后加入溴代糖8g,反应2h后再补加溴代糖2g,反应6~15h后TLC监测反应是否完全。抽滤,滤饼用二氯甲烷洗至滤液无色,加二氯甲烷360mL、水500mL萃取。分离出二氯甲烷层及水层,水层再分别用240mL、120mL二氯甲烷各萃取1次。合并3次萃取的二氯甲烷液,加水洗涤2~3次。二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,得到紫褐色溶液,再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)3.83g,收率57.2%,色谱纯度为95%以上。1HNMR(500MHz,DMSO),δ(ppm):8.11(2H, d, J=8.5HZ), 7.36(2H, s), 7.34(1H, s), 6.87(1H, d, J=2.25Hz), 6.84(1H, s), 5.93(1H, d,J=7.69Hz), 5.44(1H, t, J=9.53Hz), 5.16(1H, m), 5.11(1H, t, J=9.69Hz), 4.78(1H, d, J=9.83Hz), 3.63(3H, s), 2.31(3H, s), 2.30(3H, s), 2.02(3H, s), 2.00(6H, s)。反应方程式如图6所示。
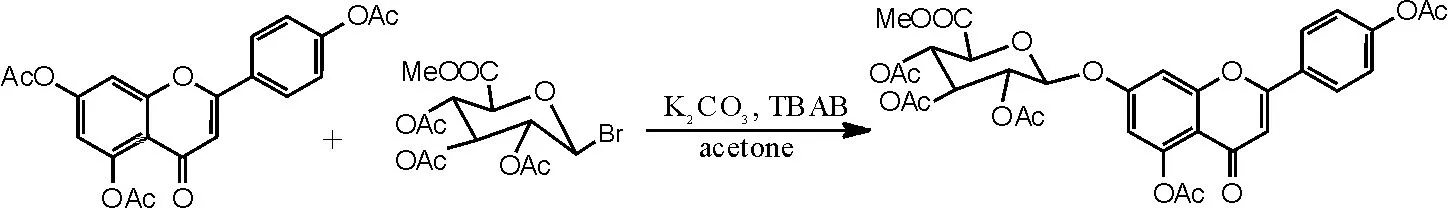
图6 相转移催化剂催化灯盏花甲素合成方程式
1.1.3 小结
化合物Ⅰ和化合物Ⅱ结构类似,前者比后者仅在6位多一个乙酰基(吸电子基团),使得化合物Ⅰ的7位乙酰基更容易水解,且水解后7位氧负离子进攻碳正离子的活性更强。因此,化合物Ⅰ比化合物Ⅱ更容易进行糖苷化,且收率更高。
(1)催化剂对糖苷化反应的影响。在糖苷化反应中,贵金属催化剂较相转移催化剂的收率提高了约5%,但其成本高昂,不适用于工业化生产。经大量实验验证,相转移催化剂的加入量为原料的0.5~2个当量,贵金属催化剂的加入量为原料的1~2个当量为最佳。
(2)受体对糖苷化反应的影响。化合物Ⅰ和化合物Ⅱ结构很相近,但接受糖配体的活性却有很大差别,最佳反应条件也有很大差异。
(3)温度对糖苷化反应的影响。最佳反应温度因受体而异,经总结发现,糖苷化反应的最佳温度为20~40℃:温度过低则反应过慢(大于24h),温度过高则促进了溴代糖、化合物Ⅰ和化合物Ⅱ的水解,不利于糖苷化反应的进行。
(4)糖苷化反应体系的最佳pH值为6~8。在相转移催化剂催化灯盏花甲素合成的糖苷化反应中,若采用与灯盏花乙素合成糖苷化反应一致的条件,反应基本不会发生,或者很慢,收率极低,约为5%~10%,继续提高催化剂量对反应速率影响较小,或基本无影响;而升高温度(温度不超过40℃)或增加碱的量可使得原料中7-OH的裸露量增多,在此种情况下,反应收率相对提高(10%~15%),但耗费的溴代糖量大大提高,因为在碱性及温度升高的情况下,溴代糖水解增加,耗费的溴代糖量也增加。溴代糖水解会产生氢溴酸,可消耗一部分碱,而在碱度不足的情况下,反应进行很慢或者基本不进行。此时若再补充碱维持原来的碱度,可勉强推进反应进程,但与此同时水解的溴代糖也随之增加。如此周而复始,不仅无法大幅度提高反应速率,反而浪费试剂且耗时。
综上所述,在相转移催化剂用于灯盏花甲素合成的糖苷化反应中,选择另一种溶剂,另一种反应条件来完成其糖苷化反应,可使得最终收率相对提高。
1.1.4 讨论
灯盏花乙素与灯盏花甲素唯一的区别为黄酮母核上的羟基数目不同,灯盏花乙素的6位多了一个羟基,二者活性最强的基团均为7位上的羟基。而糖苷化反应就是要在其7位上接一个全乙酰葡萄糖醛酸甲酯。理论上讲,由于分子间作用力、空间位阻及键能的影响,酚羟基数目越多,糖苷化反应越难进行。但从实验结果来看,在相转移催化剂的作用下,相同反应条件下,灯盏花甲素合成的糖苷化反应更难进行些;从重金属盐催化中也可以看出,灯盏花甲素合成中糖苷化反应的收率相对低于乙素,其具体原因还需要进一步探讨。
1.2 不同催化剂对以化合物Ⅶ或Ⅷ为原料的糖苷化反应的影响
以化合物Ⅶ或Ⅷ为原料进行糖苷化反应,首先是合成出化合物Ⅶ或Ⅷ。以化合物Ⅰ或Ⅱ为起始原料,需经过苄氯取代、氢化还原两步反应才能得到产物,具体实验情况如下。
1.2.1 苄氯取代,化合物Ⅴ合成
[18]反应步骤,具体操作如下:向250mL洁净干燥的三口瓶中加入化合物Ⅰ4.54g(0.01moL),70mL丙酮,待原料溶解后加入2mL氯化苄,再加入3g 碳酸钾与2g碘化钾,50~60℃下加热回流反应7~9h, HPLC监测反应是否完全(由于此反应中原料及产物极性相似,TLC监测很难辨别原料点及产物点,故选择HPLC精密监控反应进程)。待反应无原料且产物量不再增加时停止反应,减压蒸去丙酮,剩余固体加入150mL二氯甲烷后摇匀,使产物尽量完全溶解于二氯甲烷中;抽滤,滤饼用二氯甲烷洗至滤出的滤液无色,加水100mL,萃取,水层再分别用100mL、50mL二氯甲烷各萃取1次。合并3次萃取的二氯甲烷液并加水洗涤2~3次。二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,浓缩物用乙醇重结晶,抽滤,干燥,得到灰白色晶体化合物Ⅴ(7-苄氧基- 5,6,4'- 三乙酰氧基黄酮)3.35g,收率为66.7%。反应方程式如图7所示。
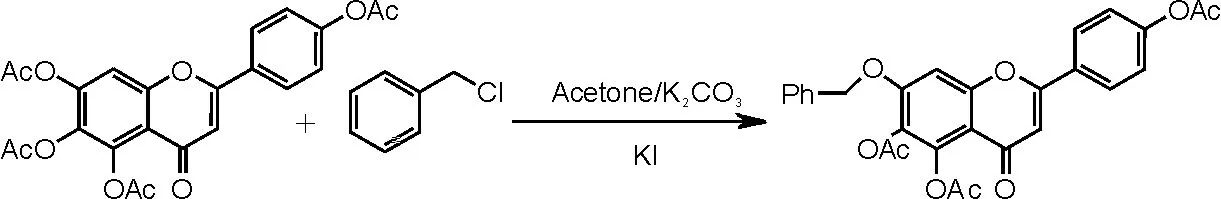
图7 化合物Ⅴ合成反应方程式
1.2.2 苄氯取代,化合物Ⅵ合成
参考文献[19]反应步骤,具体操作如下:向250mL洁净干燥的三口瓶中加入化合物Ⅱ3.96g(0.01moL)和70mL丙酮,待原料溶解后在搅拌下加入2mL氯化苄、3g 碳酸钾与2g碘化钾,50~60℃下加热回流反应6~8h,HPLC监测反应是否完全(由于此反应中原料及产物极性相似,TLC监测很难辨别原料点及产物点,故选择HPLC精密监控反应进程)。待反应无原料且产物量不再增加时,停止反应;减压蒸去丙酮,剩余固体加入150mL二氯甲烷后摇匀,使产物尽量完全溶解于二氯甲烷中;抽滤,滤饼用二氯甲烷洗至滤出的滤液无色,加水100mL,萃取,水层再分别用100mL、50mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液,加水洗涤2~3次。二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,浓缩物用乙醇重结晶,抽滤,干燥,得到灰白色化晶体化合物Ⅵ(7-苄氧基- 5,4'-二乙酰氧基黄酮)2.85g,收率为64.2%。反应方程式如图8所示。
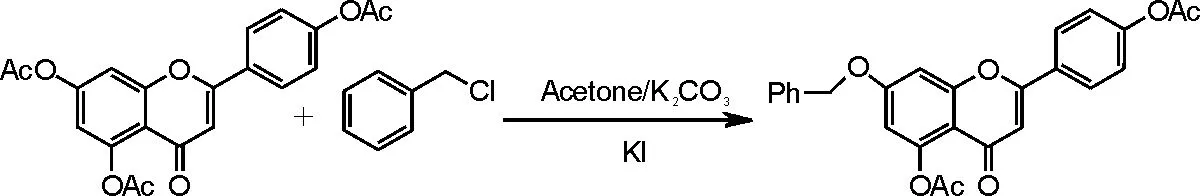
图8 化合物Ⅵ合成反应方程式
1.2.3 氢化还原,化合物Ⅶ合成
参考文献[18]反应步骤,具体操作如下:向密闭的 250mL圆底三口瓶中加入化合物Ⅴ5.02g(0.01moL)和100mL碳酸二甲酯使之溶解,再加入0.7gPd/C(10%),减压抽真空使三口瓶中无氧,再向三口瓶中通入高纯氢气,常温(20~30℃)搅拌48~52h,待反应瓶中有少量白色固体析出时停止反应,然后加热反应液回流,趁热过滤,滤液放置过夜,抽滤,干燥,得到白色晶体化合物Ⅶ(7-羟基- 5,6,4'- 三乙酰氧基黄酮)3.75g,收率为91.0%。反应方程式如图9所示。
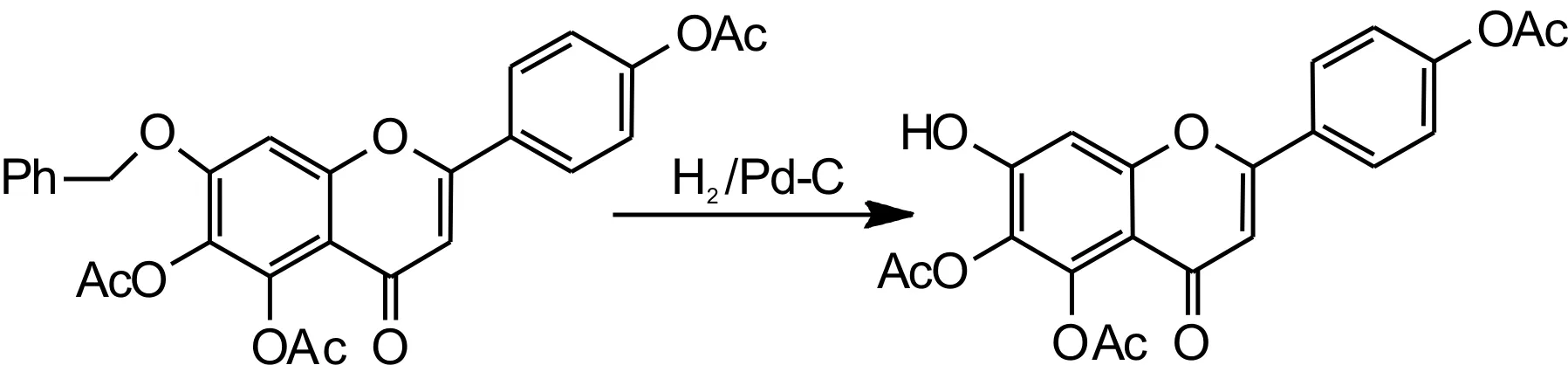
图9 化合物Ⅶ合成反应方程式
1.2.4 氢化还原,化合物Ⅶ合成
参考文献[19]反应步骤,具体操作如下:在密闭的 250mL圆底三口瓶中加入化合物Ⅵ4.44g(0.01moL)和 100mL碳酸二甲酯使之溶解,再加入0.7gPd/C(10%)。减压抽真空使三口瓶中无氧,再向三口瓶中通入高纯氢气,常温(20~30℃)搅拌48~52h,待反应瓶中有少量白色固体析出时停止反应,然后加热反应液回流,趁热过滤,滤液放置过夜,抽滤,干燥,得到白色晶体化合物Ⅷ(7-羟基- 5,6,4'- 三乙酰氧基黄酮)3.15g,收率为89.0%,反应方程式如图10所示。
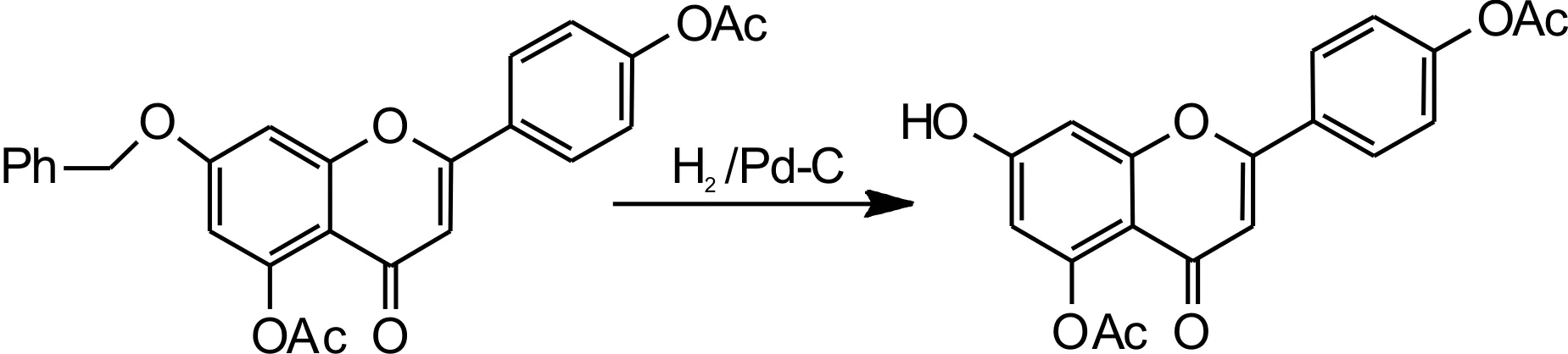
图10 化合物Ⅶ合成反应方程式
1.2.5 化合物Ⅲ和Ⅳ合成
(1)重金属盐催化化合物Ⅲ的合成。参考文献反应步骤[15], 具体操作如下:向干燥洁净的250mL圆底烧瓶中依次加入化合物 Ⅶ 4.12g(0.01moL)、喹啉60mL、无水硫酸钠9g、碳酸银3.3g、溴代糖10.3g,温度为 15~30℃,TLC跟踪监测反应,搅拌反应2h后再补加溴代糖2.1g,反应6~
15h,TLC监测反应是否完全;抽滤,滤饼用二氯甲烷洗至滤液无色,用稀盐酸溶液中和滤液至pH呈中性,除去剩余的喹啉;加二氯甲烷180mL、水200mL萃取,分离出二氯甲烷层及水层,水层再分别用120mL、60mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液加水洗涤2~3次。二氯甲烷液用无水硫酸钠干燥,再次抽滤,除去无水硫酸钠及残余的碳酸银,蒸干二氯甲烷,得到紫褐色溶液。再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅲ(5,6,4'- 三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)6.41g,收率为88.0%,色谱纯度为95%以上。1H-NMR (500MHz, DMSO), δ (ppm): 8.10 (2H, d, J=8.6 Hz), 7.53 (1H, s), 7.37 (2H, d, J= 8.6Hz), 6.88 (1H, s), 5.88 (1H, d, J= 7.8Hz), 5.47 (1H, m), 5.20 (1H, m), 5.10 (1H, m), 4.84 (1H, d, J=10Hz), 3.65 (3H, s), 2.33 (3H, s), 2.31 (3H, s), 2.27 (3H, s), 2.02 (3H, s), 2.01 (3H, s), 1.99(3H, s),反应方程式如图11所示。
(2)重金属盐催化,化合物Ⅳ的合成。参考文献[15]反应步骤,具体操作如下:向干燥洁净的250mL圆底烧瓶中依次加入化合物Ⅷ3.54g(0.01moL)、喹啉60mL、无水硫酸钠7g、碳酸银2.8g、溴代糖8.8g,反应温度为15~30℃,TLC跟踪监测反应,搅拌反应2h后再补加溴代糖1.8g,反应6~15h,TLC监测反应是否完全。抽滤,滤饼用二氯甲烷洗至滤液无色,用稀盐酸溶液中和滤液至pH呈中性,除去剩余的喹啉;加二氯甲烷180mL、水200mL萃取,分离出二氯甲烷层及水层,水层再分别用120mL、60mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液,加水洗涤2~3次,二氯甲烷液用无水硫酸钠干燥,再次抽滤,除去无水硫酸钠及残余的碳酸银,蒸干二氯甲烷,得到紫褐色溶液。再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)5.84g,收率为87.2%,色谱纯度为95%以上。1HNMR(500MHz,DMSO),δ(ppm):8.11(2H, d, J=8.5HZ), 7.36(2H, s), 7.34(1H, s), 6.87(1H, d, J=2.25Hz), 6.84(1H, s), 5.93(1H, d,J=7.69Hz), 5.44(1H, t, J=9.53Hz), 5.16(1H, m), 5.11(1H, t, J=9.69Hz), 4.78(1H, d, J=9.83Hz), 3.63(3H, s), 2.31(3H, s), 2.30(3H, s), 2.02(3H, s), 2.00(6H, s)。反应方程式如图12所示。
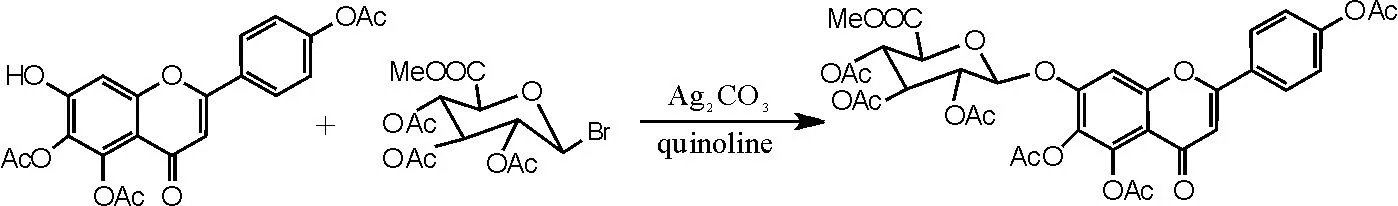
图11 化合物Ⅲ合成反应方程式
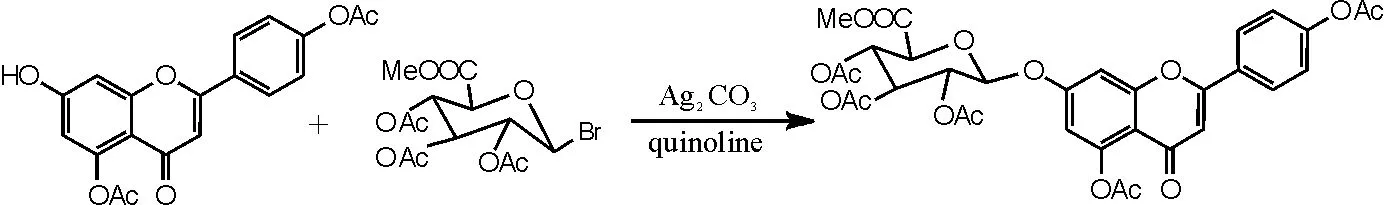
图12 化合物Ⅳ合成反应方程式
(3)相转移催化剂催化,化合物Ⅲ的合成。参考文献[16-17]反应步骤,具体操作如下:向干燥洁净的250mL的圆底烧瓶中依次加入化合物 Ⅶ 4.12g(0.01moL)、DMF 90mL、碳酸钾9g和相转移催化剂TBAB 3.5g,在磁力加热搅拌器中搅拌10min,反应温度为15~30℃,之后加入溴代糖10.0g,TLC跟踪监测反应;反应2h后再补加溴代糖2.0g,反应6~10h后TLC监测无原料。抽滤,滤饼用二氯甲烷洗至滤液无色,加二氯甲烷270mL、水400mL萃取。分离出二氯甲烷层及水层,水层再分别用180mL、90mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液加水洗涤2~3次,二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,得到紫褐色溶液。再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅲ(5,6,4'- 三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)5.47g,收率为75.1%,色谱纯度为95%以上。1H-NMR (500MHz, DMSO), δ (ppm): 8.10 (2H, d, J=8.6 Hz), 7.53 (1H, s), 7.37 (2H, d, J= 8.6Hz), 6.88 (1H, s), 5.88 (1H, d, J= 7.8Hz), 5.47 (1H, m), 5.20 (1H, m), 5.10 (1H, m), 4.84 (1H, d, J=10Hz), 3.65 (3H, s), 2.33 (3H, s), 2.31 (3H, s), 2.27 (3H, s), 2.02 (3H, s), 2.01 (3H, s), 1.99(3H, s)。反应方程式如图13所示。
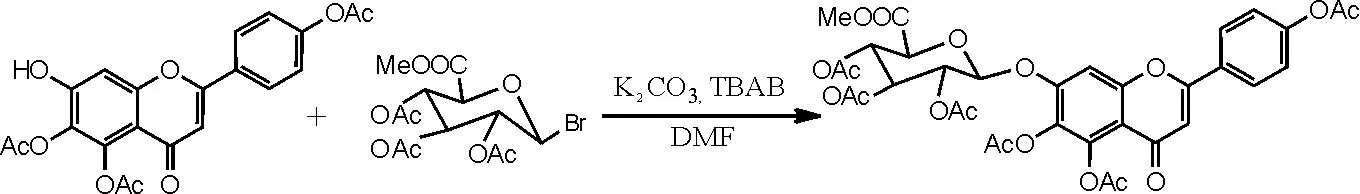
图13 化合物Ⅲ合成反应方程式
(4)相转移催化剂催化,化合物Ⅳ的合成。参考文献[16-17]反应步骤,具体操作如下:向干燥洁净的250mL圆底烧瓶中依次加化合物 Ⅷ 3.54g(0.01moL)、DMF70mL、碳酸钾7g、相转移催化剂TBAB 2.8g,在磁力加热搅拌器中搅拌10min后加入溴代糖8.8g,TLC跟踪监测反应,反应2h后再补加溴代糖1.8g,反应6~10h后TLC监测无原料。抽滤,滤饼用二氯甲烷洗至滤出液无色,加二氯甲烷210mL、水300mL萃取,分离出二氯甲烷层及水层,水层再分别用140mL、70mL二氯甲烷各萃取1次,合并3次萃取的二氯甲烷液加水洗涤2~3次,二氯甲烷液用无水硫酸钠干燥,减压蒸干二氯甲烷,得到紫褐色溶液。再用乙醇重结晶,抽滤,干燥,得到灰白色化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)4.35g,收率为65.0%,色谱纯度为95%以上。1HNMR(500MHz,DMSO),δ(ppm):8.11(2H, d, J=8.5HZ), 7.36(2H, s), 7.34(1H, s), 6.87(1H, d, J=2.25Hz), 6.84(1H, s), 5.93(1H, d,J=7.69Hz), 5.44(1H, t, J=9.53Hz), 5.16(1H, m), 5.11(1H, t, J=9.69Hz), 4.78(1H, d, J=9.83Hz), 3.63(3H, s), 2.31(3H, s), 2.30(3H, s), 2.02(3H, s), 2.00(6H, s)。反应方程式如图14所示。
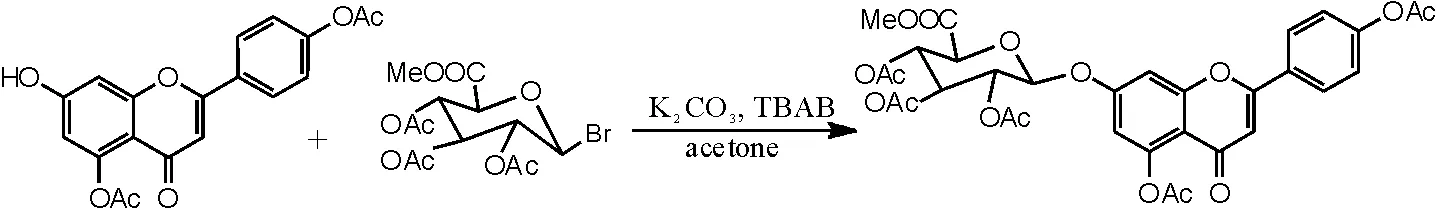
图14 化合物Ⅳ合成反应方程式
2 结语
(1)催化剂对糖苷化反应的影响。本文主要探讨贵金属催化剂与相转移催化剂对灯盏花甲素及灯盏花乙素合成中糖苷化反应的影响。在实际的糖苷化反应中,贵金属催化剂较相转移催化剂的收率高出约5%,但其成本高昂,不适用于工业化生产。经大量的实验探索,相转移催化剂的加入量为原料的0.5~2个当量,贵金属催化剂的加入量为原料的1~2个当量时反应效果最佳。常用的贵金属催化剂为碳酸银和氧化银,常用的相转移催化剂为正四丁基溴化铵、正四丁基碘化铵、三(3, 6-二氧杂庚基)胺、苄基三乙基溴化铵或三辛基甲基氯化铵等。
(2)温度对糖苷化反应的影响。最佳反应温度因受体而异,笔者总结发现,糖苷化反应的最佳温度为20~40℃,温度过低则反应过慢(大于20h),温度过高则促进了溴代糖和糖苷化受体的水解,不利于糖苷化反应的进行。
(3)pH对糖苷化反应的影响。糖苷化反应的最佳体系pH值为6~8,碱性过强会促进溴代糖和糖苷化受体水解,不利于糖苷化反应的进行。
(4)不同催化剂作用下不同受体进行糖苷化反应的收率。在相同反应条件下,化合物Ⅶ和化合物Ⅷ糖苷化反应的收率均优于化合物Ⅰ和化合物Ⅱ,但从化合物Ⅰ(Ⅱ)到化合物Ⅶ(Ⅷ)尚需经过苄氯取代和氢化还原两步反应,使其糖苷化反应的总收率大大降低,不适合工业化生产。
不同催化剂作用下不同受体进行糖苷化反应收率如下所示。
(1)在不同催化剂作用下直接以化合物Ⅰ或Ⅱ所进行糖苷化反应的收率如下。重金属盐催化:合成化合物Ⅲ(5,6,4'- 三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)的收率约为67.3%;合成化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)的收率约为63.15%。相转移催化剂催化:合成化合物Ⅲ(5,6,4'- 三乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)的收率约为61.1%;合成化合物Ⅳ(5,4'- 二乙酰氧基黄酮-7-0-D-三乙酰氧基葡萄糖醛酸甲酯)的收率约为57.2%。
(2)在不同催化剂作用下化合物Ⅶ或Ⅷ为原料进行糖苷化反应的收率如下。重金属盐催化:最终合成化合物Ⅲ的总收率=苄氯取代收率×氢化还原收率×糖苷化收率=66.7%×91.0%×88.0%≈53.4%,最终合成化合物Ⅳ的总收率=苄氯取代收率×氢化还原收率×糖苷化收率= 64.2%×89.0%×87.2%≈51.4%。相转移催化剂催化:最终合成化合物Ⅲ的总收率=苄氯取代收率×氢化还原收率×糖苷化收率=66.7%×91.0%×75.1%≈45.6%,最终合成化合物Ⅳ的总收率=苄氯取代收率×氢化还原收率×糖苷化收率= 64.2%×89.0%×65.0%≈37.1%。
3 结语
化合物Ⅰ和化合物Ⅱ结构类似,前者比后者仅在6位多一个乙酰基(吸电子基团),这使得化合物Ⅰ的7位乙酰基更容易水解,且水解后7位氧负离子进攻碳正离子的活性更强。因此,化合物Ⅰ(Ⅶ)比化合物Ⅱ(Ⅷ)更容易进行糖苷化反应,且收率更高。贵金属盐催化的糖苷化反应收率高于相转移催化剂,但成本较高,环境污染更大,且后处理繁琐,不利于工业化生产。若不经过7位的苄基取代和选择性还原,不管是重金属盐催化还是相转移催化剂催化,以化合物Ⅰ和化合物Ⅱ直接进行糖苷化的总收率都更高。本文所述的相转移催化反应直接以全乙酰化野黄芩素或芹菜素为原料进行糖苷化反应,具备了工业化生产的条件,具有潜在的药用价值、经济价值。若要开发出更合适的催化剂及收率更高的合成方法,如酶催化的生物合成法等,还需要广大科研工作者继续努力。
参考文献:
[1] 张卫东,陈万生,孔德云,等.灯盏细辛化学成分的研究[J].中国药学杂志,2000(8): 10-12.
[2] 张人伟,张元玲,王杰生,等.灯盏花黄酮类成分的分离鉴定[J].中草药,1988,19(5):71.
[3] 周建中,雷寒,陈运贞,等.灯盏细辛注射对自发性高血压大鼠心室及血管熏构的影响[J].中国中西医结合杂志,2002,22(2):122-125.
[4] 盛净,赵佩琪,黄震华,等.灯盏细辛干预血小板、凝血功能对急性冠状动脉血栓形成后溶栓的影响[J].中华心血管病杂志,1999,27(2):115-116.
[5] 张永和,宋祖军,张新睿.灯盏花乙素对脑梗死患者超氧化物歧化酶及丙二醛含量的影响[J].中国急救医学,2003,23(6):394-395.
[6] 王丽娟,王勇,李金鸣.灯盏花乙素对豚鼠心室肌细胞迟发性外向钾电流的影响[J].中国药理学通报,2002,18(3):326-325.
[7] 张焰,陈群,丁浩中,等.灯盏花乙素注射液对脑缺血再灌注沙土鼠海马ATP含量和ATP酶活性变化的影响[J].中国中医结合急救杂志,2002,9(2): 92-94.
[8] 姜瑞芝,高其品,唯大员,等.芹菜素-7-O-B-D葡萄糖醛酸营在制备治疗痴呆疾病药物中的作用[P].中国:CN:200810050872.3,2008-10-29.
[9] MIN Y,YIM S,BAI K,et al.The effeets of aPigenin-7-O-B-D-glueuronoPyra noside on reflux oesoPhagitis and gastritis in rats[J].Autonomic & Autacoid Pharmacology,2005,25(3):85-91.
[10] WAGNER H,DANNINGER H,SELIGMANN,et al. Synthesis of glucuronides in the flavonoid series part 5 the 1st synthesis of a naturally occurring flavonoid di glucuronide apigenin 4 7 di-o-beta-d glucuronide and the synthesis of chrysoeriol 7 mono-o-beta-d glucuronide[J]. Chemische berichte, 1973,106(8): 2536-2541.
[11] DEMETZOS C, SKALTSOUNIS A L, RAZANAMAHEFA B, et al. Synthesis of quercetin-3-O-beta-D-glucopyranosyl-(1->2)-beta-D- xylopyranoside via ortho-ester methodology[J]. J Nat Prod,1994,57(9): 1234-1238.
[12] S T CALDWELL, A CROZIER, R C HARTLEY. Isotopic labeling of quercetin 4 0 -O-b-D-glucoside[J].Tetrahedron,2000(56):4101-4106.
[13] ELLY SMITS, JAM,B F ENGBERTS N,RICHARD M,et al.Reliable methods for the synthesis of aryl-B-D-Glucopyranosides, using boron trifluoride-diether as catalyst[J].J.Chem.Soc.,Perkin Trans.I,1996(24):2873.
[14] VIE G, CRONTD H G.Synthesis of allyl and benzyl β-d-glucopyranosides and allyl β-d-galactopyranoside from d-glucose or d-galactose and the corresponding alcohol using almond β-d-glucosidase[J].Carbohydr.Res,1995(279):315-319.
[15] 蒋忠良,朱胃远,伍越环,等.黄酮类天然产物的全合成研究[J].药学学报,1994,29(11):874-876.
[16] 陈贺.生理活性黄酮及7-O-糖基黄酮苷类化合物的合成研究[D].长沙:湖南大学,2006.
[17] WANG YAPING,LI LIANGXI,WANG OINGLIAN. An improved phase transfer catalyzed synthetic Method for onion and rothindin[J].Synth. Commun,2001,31(22):3423-3427.
[18] 陈铎之.灯盏花乙素合成研究[D].昆明:昆明理工大学,2009.
[19] 吴婷.天然黄酮苷灯盏花甲素合成研究[D].昆明:昆明理工大学,2011.
(责任编辑:尹晨茹)
Study of Scutellarin and Scutellarin Biosynthesis in Glycosidation Reaction
Zhang Zhipeng1, Yang Zhaoxiang2, Li Penghui2, Zhang Wei2
(1.The Kunming Medical University School of medicine, Yunnan 650500,China;2.Kunming Pharmaceutical Group Pharmaceutical Research Institute, Yunnan 650106,China)
breviscapine are a class of flavonoids extracted from Erigeron breviscapus its main components include scutellarin and apigenin-7-O-β-D-glucuronide . Scutellarin is the main active ingredient of Erigeron breviscapus drugs, the synthesis method of apigenin-7-O-β-D-glucuronide and pharmacological studies reported in the literature are few. The key steps of scutellarin and apigenin-7-O-β-D-glucuronide synthesis process is the glycosidation, traditional scutellarin and apigenin-7-O-β-D-glucuronide glycosidation reaction route is long, cost is high, is not conducive to industrial production. The glycosylation of scutellarin and apigenin-7-O-β-D-glucuronide synthesis in a large number of research, to optimize the synthetic process. The glycosylation methods by test card, can be used for industry.
Scutellarin; Apigenin-7-O-β-D-glucuronide; Glycosidation; Synthesis
2014-03-25
张志朋(1987-),男,昆明医科大学硕士研究生,研究方向为药物合成及药理学研究。
R284.3
A
1673-2197(2014)10-0016-07
猜你喜欢
中草药(2022年9期)2022-05-06
化学工程师(2022年3期)2022-04-19
上海化工(2021年2期)2021-04-23
铜仁学院学报(2018年6期)2018-07-05
中国洗涤用品工业(2016年2期)2016-02-28
中国马铃薯(2015年5期)2016-01-09
科技与企业(2015年20期)2015-10-21
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
现代检验医学杂志(2014年4期)2014-02-02
