
基于连接酶—ELISA反应的单核苷酸多态性分型新方法
2014-03-23 02:20崔海忠张永平陈大贵唐一通赵雪红邵金辉
生物学杂志 2014年4期
崔海忠, 肖 娜, 张永平, 陈大贵, 唐一通, 赵雪红, 邵金辉
(1. 湖北文理学院医学院 枣阳临床学院, 枣阳 441200; 2. 湖北文理学院医学院 分子医学重点实验室, 襄阳 441053)
单核苷酸的多态性(single nucleotide polymorphism, SNP)是最为常见的一种遗传多态性,占现有多态性的的90%以上。现有公共数据库中已经报道了超过900万个SNP位点[1]。开展SNP研究对医学、法医学、药物开发与合理用药、遗传学、临床诊断等的发展均有非常重要的意义[2]。SNP分型检测工作,已成为当前国际上研究的热点。近年来,SNP检测方法和相关仪器的研发进展很快。目前,有20余种的SNP分型方法,如直接测序,PCR-RFLP[3-4],PCR-SSCP[5], Real-Time PCR[6-7], TaqMan PCR[8], Allele-Specific Extension[9-10], Pyrosequencing[11-12], Mass Spectrometry[13-14], Ligase Detection Reaction[15-17]等。其中,直接测序法是传统的序列测定的方法,但是由于检测灵敏度较低,在对不均一样本进行突变等位基因测定时具有较大局限性。虽然焦磷酸测序、荧光定量PCR以及质谱等方法具有较高的检测灵敏度,但是由于这些方法对实验设备和实验条件要求很高,因此很少应用于一般实验条件下的常规临床检测。本研究中建立了一种基于连接酶-ELISA的SNP分型新方法,并以在非小细胞肺癌酪氨酸激酶抑制剂药物个体化治疗中的生物标记基因表皮生长因子受体(EGFR)为检测对象,对有较高突变频率的EGFR基因外显子21和18的EGFR, c.2573T>G(L858R), EGFR, c.2582T>A(L861Q) 和 EGFR, c.2155 G>T(G719C)3个SNP位点进行了检测。
1 材料和方法
1.1 实验材料
质粒模板:携带有野生型和突变型等位基因的EGFR, c.2573T>G(L858R), EGFR, c.2582T>A(L861Q) 和 EGFR, c.2155 G>T(G719C)3个SNP位点的质粒DNA。以黄杰[18]定点突变技术进行构建。

图1 检测探针设计示意图。“H1”, “H2”, “Tag1”, “Tag2”代表探针的不同组成部分。
寡核苷酸检测探针:探针设计方法如图1所示,利用3条寡核苷酸探针对每个SNP位点进行检测。3条探针分别为上游的两条特异性检测探针和下游的一条公用探针,两条特异性探针的5′端序列为一段通用扩增引物序列Tag1,3′端序列与模板上SNP位点一侧的序列互补,且3′端最后一个碱基与突变位点所在碱基对应互补,与野生型等位基因互补的为野生型检测探针(Wild Type-Probe),与突变型等位基因互补的为突变型检测探针(Mutation Type-Probe),且特异性探针的5′端进行生物素修饰。公用探针的5′端序列与SNP位点另一侧序列互补,3′端序列为一段通用扩增引物序列Tag2。 对EGFR, c.2573T>G(L858R), EGFR, c.2582T>A(L861Q) 和 EGFR, c.2155 G>T(G719C)3个SNP位点进行检测的探针序列如表1所示。

表1 寡核苷酸检测探针序列
实验试剂:2×PCR Mastermix购自天根生化科技北京有限公司,Low MW DNA Marker-A购自生工生物工程(上海)有限公司,TaqDNA ligase购自New England BioLabs,辣根过氧化物酶标记兔抗地高辛抗体购自北京博奥森生物技术有限公司,Streptavidin 磁珠购自西安金磁纳米生物技术有限公司。TaKaRaExTaq酶、TA 克隆试剂盒为宝生物工程( 大连) 有限公司产品。所有核酸序列均合成自上海生工生物有限公司。其余试剂均为分析纯。
1.2 实验方法
1.2.1 连接和扩增反应
连接反应:每个SNP检测位点对应两个反应管,分别标记为WT、MT,分别加入5 fmol 的重组质粒DNA模板序列、1 U的Taqligase和3 μL 10×连接反应缓冲液,WT管中对应加入50 fmol野生型特异性检测探针和公用检测探针,MT管中加入50 fmol突变型特异性检测探针和公用检测探针,去离子水补足30 μL。针对每个检测位点设定一对照管CT,对照管不加入模板序列,其它成分与检测管相同。连接反应条件为:94℃ 40 s,55℃ 5 min;5 cycles;95℃ 5 min。
通用扩增反应:取2×PCR Mastermix 15 μL,各管连接反应产物2 μL,2 μM的通用扩增引物Tag1 和CTag2,去离子水补足30 μL。95℃ 30 s;95℃ 25 s,60℃ 45 s,72℃ 20 s;20 cycles, 72℃ 1 min进行扩增反应。
1.2.2 扩增产物检测
琼脂糖凝胶电泳检测:分别取各SNP位点对应WT管和MT管扩增产物5 μL进行3.5%琼脂糖凝胶电泳,根据WT管和MT管对应泳道在目标位置电泳条带出现情况判定SNP位点基因型。如果只有WT管对应泳道出现条带,则该SNP位点为纯合野生型;如果WT管和MT管对应泳道均出现条带,则该SNP位点为野生/突变型;如果只有MT管对应泳道出现条带,则检测SNP为纯合突变型。
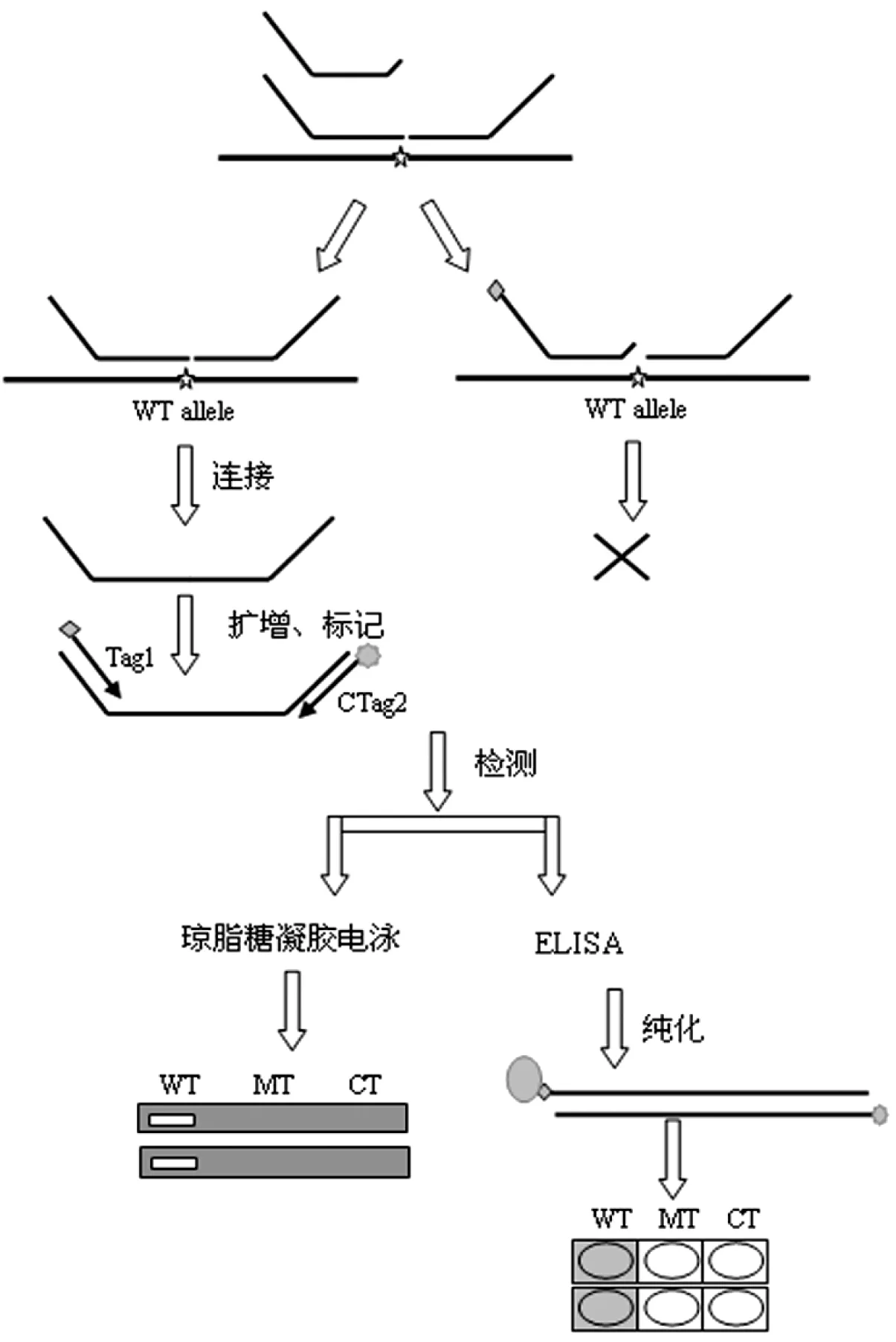
图2 连接酶-ELISA分型方法检测流程图。“” 表示生物素标记;

ELISA检测:将扩增产物移入96孔板对应孔中,按照试剂盒操作说明,用链亲和素磁性微粒对各SNP位点对应各管扩增产物进行纯化。并用PBST清洗缓冲液清洗3次磁性微粒,地高辛标记的扩增产物经纯化后通过生物素结合至链亲和素磁性微粒表面,将磁性微粒保存于20 μL 保存缓冲液(10 mM Tris-HCl, pH值7.5)中。各反应孔中加入50 μL辣根过氧化物酶标记抗地高辛抗体溶液(20%胎牛血清1∶1000 稀释),室温恒温摇床中70 r/min反应30 min。磁性分离并用150 μL清洗缓冲液(1×PBST)清洗3次。加入100 μL的TMB底物缓冲液,25℃显色 5 min。2 M H2SO4终止显色反应,设定检测波长450 nm,背景波长595 nm,酶标仪(ELX 800 UV, BIO-TEK INSTRUMENTS, INC)测定各孔吸光度值。根据各SNP位点WT管和MT管对应各孔显色值判定所检测SNP位点基因型,判定结果同琼脂糖凝胶电泳方法。
2 结果
2.1 检测方法流程
图2所示为对SNP(T/G)位点的野生纯合子进行检测的示意图,在连接反应时,只有WT管中的特异性检测探针和公用探针能完成连接,而MT管中特异性探针和公用探针不能连接,经通用引物Tag1 和CTag2的通用扩增和标记,可分别在琼脂糖凝胶电泳水平和ELISA水平进行分型检测,检测结果可通过观察WT管和MT管对应电泳泳道条带的出现和显色孔显色值来判定。如示意图中,只在WT管对应泳道出现条带,而MT管对应泳道无条带;同理,只在WT管对应显色孔有显色值,而MT管对应泳道无显色值。
2.2 特异性检测
以扩增反应中的循环数对本方法的特异性进行评估,分别对L861Q 和G719C两个野生型等位基因模板和L858R野生/突变混合模板进行检测。分别扩增18 和28个循环,将5 μL各扩增产物做3.5%琼脂糖凝胶电泳。结果如图3所示。在两个循环条件下,L858R位点的WT和MT管对应泳道均出现明显扩增条带,L861Q 和G719C两个位点只在WT管对应泳道出现扩增条带,而MT泳道未有条带扩增。同时3个位点CT管对应泳道均未出现扩增条带。说明此方法具有较高的扩增容量和检测特异性。
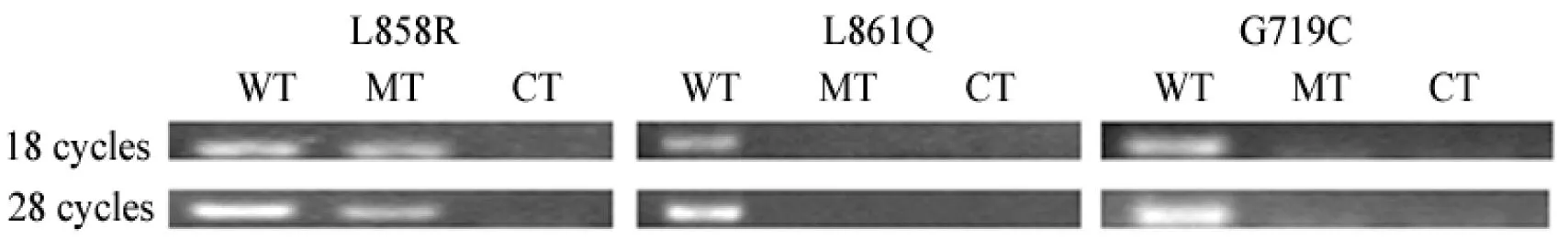
图3 连接酶-ELISA分型方法特异性检测
2.3 灵敏度检测
选取L858R位点为检测对象,突变等位基因在总检测模板中所占比例分别设定为50%、25%、10%和5%。对不同比例的突变型等位基因进行检测。
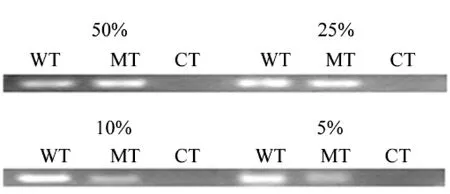
图 4 L858R位点琼脂糖凝胶电泳水平敏感度检测(突变等位基因在总检测质粒模板中所占比例分别设定为50%, 25%, 10%, 5%)
Fig 4 Sensitivity for the detection of L858R performed with 3.5% agarose gel electrophoresis (The proportions of mutated alleles in the total plasmid DNA were 50%, 25%, 10%, 5%)
结果如图4所示,在MT和WT管对应的泳道中均检测出条带,且随着突变型等位基因含量的减少,MT管对应条带逐渐减弱。当低至5%时条带较为模糊。为了提高方法的灵敏度,将ELISA方法引入实验体系中,结果如表2所示。L858R, L861Q 和 G719C位点对应WT管的450 nm处的光吸收值在1.68和1.89之间,MT管的450 nm处的光吸收值在0.71和1.86之间。L858R, L861Q和G719C 3个位点MT管在突变等位基因比例为5%时的光吸收值分别达到0.81、0.71 和0.76。而且,WT和MT管对应光吸收与CT管对应光吸收值的比值均大于5。因此,ELISA水平的检测有着较高的灵敏度,能够准确的将5%的突变等位基因从混合模板中检测出来,尤其适合于从不均一的样本中进行突变等位基因的检测。
表2 L858R、L861Q和G719C位点ELISA水平敏感度检测(突变等位基因在总检测质粒模板中所占比例分别设定为50%, 25%, 10%, 5%,各检测值为三次检测平均值± S.E)
Table 2 Sensitivity for the detection of L858R、L861Q and G719C in exon21 and 18 of EGFR with the ELISA (The proportion of mutated allele in the total plasmid DNA was 50%, 25%, 10%, 5% respectively, values of each reaction tube are means ± S.E. obtained from three independent experiments)

突变等位基因在总检测质粒模板中所占的比例50%25%10%5%WTL858R1.76±0.111.81±0.151.78±0.171.82±0.14G719C1.82±0.131.79±0.121.86±0.091.78±0.18L861Q1.83±0.161.68±0.211.89±0.111.79±0.08MTL858R1.85±0.091.44±0.161.01±0.180.81±0.09G719C1.72±0.121.68±0.111.09±0.20.76±0.2L861Q1.86±0.141.12±0.130.95±0.130.71±0.05CTL858R0.15±0.020.13±0.030.14±0.020.09±0.02G719C0.12±0.030.09±0.060.11±0.050.07±0.04L861Q0.14±0.020.07±0.070.08±0.040.08±0.02
3 讨论
随着人类基因组计划的完成,使得生物信息学资源获得了爆炸性的积累,也极大地推动了SNP突变检测技术的发展。但是,虽然在许多技术上取得了进展,SNP检测技术在实验成本、常规性和灵敏度等方面仍需要改进。由于传统的测序方法检测灵敏度只有20%[19-20],不适合对不均一样本进行突变检测,虽然焦磷酸测序、定量PCR、质谱等方法的检测灵敏度较高,可分别达到5%和1%[20-21],但是这些方法都要求昂贵的实验试剂和检测设备,不利于在常规实验条件下进行检测。
本研究中,建立了一种具有较高检测灵敏度适合于对不均一样本进行SNP突变检测而且能够在一般实验条件下进行常规突变检测的新方法:连接酶-凝胶-ELISA SNP分型方法。本方法基于连接酶反应的原理,通过连接和扩增反应,利用琼脂糖凝胶电泳和ELISA检测手段进行SNP位点的分型检测。与其它检测方法如测序、荧光定量PCR反应和质谱检测等相比,本方法操作简单,不需要昂贵的试剂和实验仪器,只需借助PCR仪、凝胶电泳和酶标仪等简单实验条件下就能完成。
本方法具有较高的特异性和灵敏度。其特异性通过以下条件保证:1)高保真性Taqligase的识别能力;2)检测探针的设计方法,除依靠WT和MT探针3′端碱基与突变碱基的配对识别能力外,还在其3′端上游第3个碱基处引入一个错配碱基增强探针的识别能力;3)WT、MT探针和公用探针进行连接反应的H1和H2序列的Tm值与进行通用扩增反应的Tag1和Tag2序列的Tm值相差7~8℃,以减少探针的连接反应和后续的扩增反应之间的影响。而本方法较高的检测灵敏度则是通过在探针序列上设计通用扩增序列Tag1和Tag2,完成连接酶反应后,在琼脂糖凝胶电泳水平检测时,进行了连接产物的二次扩增反应以增加检测量;在ELISA水平检测时,完成二次扩增产物的固相纯化后,利用显色反应进行检测。
总之,本研究建立了一种SNP分型新方法,并对与酪氨酸激酶抑制剂用药密切相关的表皮生长因子基因EGFR中的3个常见SNP位点进行了检测。与现有方法相比,该方法具有较高检测特异性和灵敏度,适合于在一般实验条件下对不均一样本进行常规SNP分型检测。但本方法不能对未知SNP位点进行突变检测,在检测通量上有一定的局限,适合于进行中低通量的检测,而不利于进行高通量的突变检测。
参考文献:
[1]Rocha D, Gut I, Jeffreys A J, et al. Seventh international meeting on single nucleotide polymorphism and complex genome analysis: “ever bigger scans and an increasingly variable genome"[J]. Hum Genet, 2006, 119:451-456.
[2]Kathryn M M. Factors influencing warfarin dose requirements in African-Americans[J]. Phamacogenomics, 2007, 8(11):1535-1545.
[3]Qiao W, Wang T, Zhang L, et al. Association study of single nucleotide polymorphisms in XRCC1 gene with the risk of gastric cancer in chinese population[J]. Int J Biol Sci, 2013, 9(7):753-758.
[4]Senol Tuncay S, Okyay P, Bardakci F. Identification of NF-kappaB1 and NF-kappaBI Alpha polymorphisms using PCR-RFLP assay in a Turkish population[J]. Biochem Genet, 2010, 48(1/2):104-112.
[5]Serrano M L, Yunis J J. Identification of three new mutations in the RB1 gene in patients with sporadic retinoblastoma in Colombia[J]. Biomedica, 2013, 33(1):53-61.
[6]Hung C C, Chiou M H, Huang B H, et al. Impact of genetic polymorphisms in ABCB1, CYP2B6, OPRM1, ANKK1 and DRD2 genes on methadone therapy in Han Chinese patients[J]. Pharmacogenomics, 2011, 12(11):1525-1533.
[7]Azzari C, Moriondo M, Indolfi G, et al. Real time PCR is more sensitive than multiplex PCR for diagnosis and serotyping in children with culture negative pneumococcal invasive disease[J]. PLoS One, 2010, 5(2):e9282.
[8]Farivar T N, Nezam M K, Johari P. Genotyping of hepatitis C virus isolated from hepatitis patients in Southeast of Iran by Taqman Realtime PCR[J]. J Pak Med Assoc, 2011, 61(6):586-588.
[9]Cai Y, Yuan Y, Lin Q, et al. Allele-specific extension allows base-pair neutral homozygotes to be discriminated by high-resolution melting of small amplicons[J]. Anal Biochem, 2010, 406(1):29-33.
[10]Liu Y H, Chen C C, Liao L L, et al. Association of IL12B polymorphisms with susceptibility to Graves ophthalmopathy in a Taiwan Chinese population[J]. J Biomed Sci, 2012, 19:97.
[11]Kang S H, Pyo J Y, Yang S W, et al. Detection of BRAF V600E mutation with thyroid tissue using pyrosequencing: comparison with PNA-clamping and real-time PCR[J]. Am J Clin Pathol, 2013, 139(6):759-764.
[12]Altimari A, De Biase D, De Maglio G, et al. 454 next generation-sequencing outperforms allele-specific PCR, Sanger sequencing, and pyrosequencing for routine KRAS mutation analysis of formalin-fixed, paraffin-embedded samples[J]. Onco Targets Ther, 2013(6):1057-1064.
[13]Mao Y, Tan F, Yan S G, et al. High-throughput genotyping of single-nucleotide polymorphisms in ace-1 gene of mosquitoes using MALDI-TOF mass spectrometry[J]. Insect Sci, 2013, 20(2):167-174.
[14]Mauger F, Gelfand D H, Gupta A, et al. High-specificity single-tube multiplex genotyping using Ribo-PAP PCR, tag primers, alkali cleavage of RNA/DNA chimeras and MALDI-TOF MS[J]. Hum Mutat, 2013, 34(1):266-273.
[15]Xu G, You Q, Pickerill S, et al. Application of PCR-LDR-nucleic acid detection strip in detection of YMDD mutation in hepatitis B patients treated with lamivudine[J]. J Med Virol, 2010, 82(7):1143-1149.
[16]Pingle M, Rundell M, Das S, et al. PCR/LDR/universal array platforms for the diagnosis of infectious disease[J]. Methods Mol Biol, 2010, 632:141-157.
[17]Yi P, Chen Z, Yu L, et al. Development of a PCR/LDR/capillary electrophoresis assay with potential for the detection of a beta-thalassemia fetal mutation in maternal plasma[J]. J Matern Fetal Neonatal Med, 2010, 23(8):920-927.
[18]黄 杰, 曲守方, 徐 任, 等. 人类EGFR基因突变体质控品的建立[J]. 药物分析杂志, 2011, 31(9):1758-1763.
[19]Garcia C A, Ahmadian A, Gharizadeh B, et al. Mutation detection by pyrosequencing: sequencing of exons 5-8 of the p53 tumor suppressor gene[J]. Gene, 2000, 253:249-257.
[20]Dufort S, Richard M J, de Fraipont F. Pyrosequencing method to detect KRAS mutation in formalin-fixed and paraffin-embedded tumor tissues[J]. Anal Biochem, 2009, 391:166-168.
[21]Jarry A, Masson D, Cassagnau E, et al. Real-time allele-specific amplification for sensitive detection of the BRAF mutation V600E[J]. Mol Cell Probes, 2004, 18(5):349-352.
猜你喜欢
智慧健康(2021年17期)2021-07-30
国际检验医学杂志(2021年7期)2021-04-15
数学大王·低年级(2020年8期)2020-08-14
中国产前诊断杂志(电子版)(2020年1期)2020-05-21
生物工程学报(2019年1期)2019-01-30
现代检验医学杂志(2016年5期)2016-08-20
小雪花·初中高分作文(2016年9期)2016-05-14
系统工程与电子技术(2016年2期)2016-04-16
中国光学(2015年1期)2015-06-06
海岸工程(2014年4期)2014-02-27
