
糖皮质激素作用机制的研究进展
2014-03-21 07:09综述刘志红审校
肾脏病与透析肾移植杂志 2014年5期
刘 琳 综述 刘志红 审校
糖皮质激素因其显著的抗炎和免疫抑制作用广泛应用于临床治疗过敏、哮喘、自身免疫病和脓毒症等疾病。在肾脏疾病中,糖皮质激素通常被用于治疗肾病综合征。在基因调控层面,糖皮质激素通过糖皮质激素受体(GR)发挥其生理及药理作用。GR作为核受体家族的一员,通过与DNA反应元件直接结合或与其他转录因子相互作用促进或抑制靶基因的转录。在细胞功能层面,激素通过抑制体液及细胞免疫治疗蛋白尿性肾脏疾病及自身免疫性疾病。而近年来糖皮质激素已经被证实对足细胞有直接保护作用。概括说来,糖皮质激素不仅能够通过抗炎和免疫抑制作用间接保护肾脏,而且能通过抑制足细胞凋亡、维持足细胞形态等对足细胞起到直接保护作用,这些发现为深入研究糖皮质激素对蛋白尿性肾脏疾病的治疗机制及激素抵抗打下了良好的基础。
GR信号通路
GR是配体依赖转录因子中核受体超家族的一员,糖皮质激素易于通过细胞膜进入细胞,与胞质内GR结合从而发挥重要的生理和药理作用。10%~20%的人类基因均能受到GR的调控从而其转录被诱导或抑制[1,2]。细胞对激素的反应在特异性和敏感性上均呈显著多样性,如激素能杀伤胸腺细胞和成骨细胞,却能保护肝细胞、足细胞和心肌细胞。而且,对激素反应的敏感性体现在不同个体、同一个体的不同器官组织、甚至在同一细胞内细胞周期的不同阶段[3]。
经典GR信号通路GR由三个主要结构域构成:氨基端的反式激活区(NTD)、中心的DNA结合区(DBD)和羧基端的配体结合区(CTD)。与激素结合后,GR的构象发生改变,蛋白复合物被分解,GR迅速通过核孔转运至细胞核内,与糖皮质激素反应元件(GRE)结合,并调控靶基因(图1)。GGAACAnnnTGTTCT是公认的GRE,包括两个6 bp的半臂及3 bp的间隔。GR能以二聚体的形式与6b的半臂结合。除GRE,一个负性GRE(nGRE)也被发现能够介导激素依赖的特殊基因转录抑制,其序列为CTCC(n)0-2GGAGA,与GRE的两臂序列不同,且间隔了一个0~2 bp的可变区域,使其只能与GR单体结合而不能形成二聚体[4]。关于nGRE还需更多的证据支持其是激素直接抑制基因的结合位点,同时也需要考虑其是否能诱导某些基因的转录。
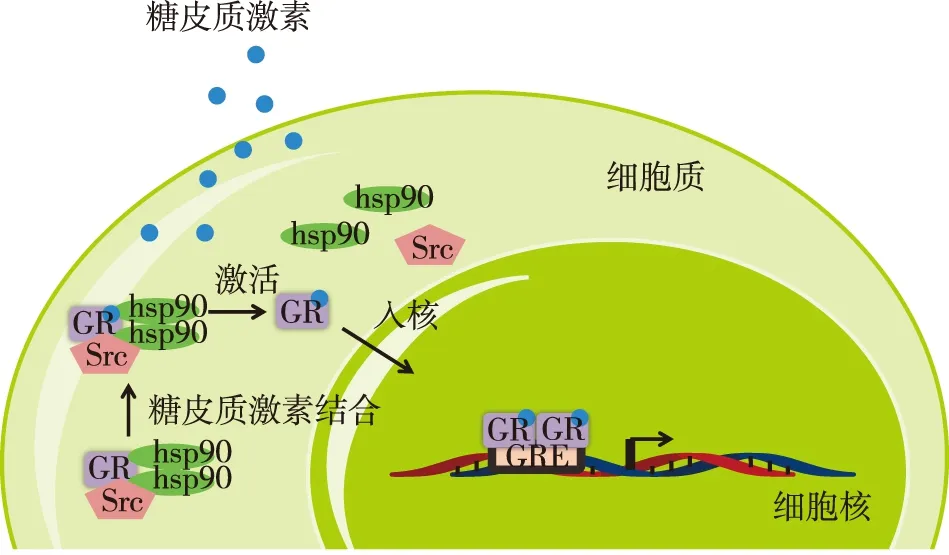
图1 糖皮质激素在细胞内的转运

图2 糖皮质激素受体信号通路[7]
John等[5]发现基因组中只有小部分GRE与受体结合,由于染色质的结构差异和GRE的暴露的区别,GR结合的特定位点有组织特异性。这些发现提示糖皮质激素在不同组织的表现多样性能部分归结于染色质环境的细胞特异性,即GRE是否向GR开放。全基因组分析也发现大部分的GR结合位点在激素反应性基因的启动子区之外,而存在于远离转录起始位点的基因间或基因内。在GR基因的第六外显子的基因内nGRE 介导了同源的GR表达下调,可能是导致糖皮质激素治疗反应差的一个重要机制[6]。GRE和nGRE存在于转录起始位点的远端提示了这些反应元件能通过形成远距离环路影响靶基因的启动子区,从而调控基因的转录。
GR与DNA的结合是高度动态的,一旦与GRE结合,GR的构象随即发生改变,能招募共转录因子,如激素受体共刺激因子SRC1-3和组蛋白乙酰化转移酶CBP/p300,从而诱导基因转录。另一方面,GR与nGRE结合能招募共抑制因子,如核受体共抑制因子N-CoR和SMRT,从而抑制基因转录[7]。GR也能通过其他转录因子的相互作用调控某些基因的转录。GR与STAT家族中特异成员相互作用能增强反应性基因的转录。相反的GR与促炎转录因子AP1和NF-κB的结合能拮抗其活性,这一作用被认为是激素抗炎反应的重要机制(图2)。
非经典的GR信号通路虽然GR的主要作用是在数分钟至数小时内通过在转录水平对靶基因进行调控,越来越多的证据表明GR也能在数秒至数分钟内通过非基因机制引起快速的细胞反应,而不发生基因表达的变化[8]。糖皮质激素能直接作用于细胞膜的双层磷脂膜结构,通过作用于细胞膜上的钙离子通道或钠离子通道改变细胞的电生理。在多种非基因机制中,信号传导最终会影响到多种激酶的活性,如PI3K、AKT和MAPKs。如非受体酪氨酸激酶c-Src可介导激素的非基因机制。当与GR复合物分离时,c-Src能激活多种激酶级联反应,导致膜联蛋白1的磷酸化、抑制胞质磷脂酶A2的活性以及减少花生四烯酸的释放[9]。非基因信号通路为糖皮质激素的作用机制增加了更多的复杂性和多样性,同时也为糖皮质激素在临床的个体化治疗上提出了新的解释。
糖皮质激素的免疫抑制作用
糖皮质激素通过各种不同的分子机制影响免疫细胞的存活、活性及一些炎症生物标志物的表达。
T细胞研究发现T细胞亚群的比例在肾病综合征患者发生了改变,表现为CD4+T细胞比例减少,CD8+T细胞和自然杀伤细胞比例增加。在肾病综合征的动物模型中,CD4+T细胞缺失能诱导CD8+T细胞数量的增加,并使病情恶化;而CD8+T细胞缺失能减轻肾脏损伤,说明CD4+T细胞在肾脏中起到保护作用,而CD8+T细胞则表现为有害作用[10,11]。
激素通过非基因机制与T细胞膜表面受体(TCR)结合,通过抑制蛋白激酶 Scr,使其从TCR上脱落,抑制TCR信号通路下游的一些重要分子,如MAPK、JNK、PKB、PKC和p38的磷酸化,快速对TCR介导的信号传导通路产生抑制作用[12]。Harr等[13]发现,低剂量的地塞米松(1~10 nmol/L)能诱导幼稚T细胞钙信号的重组,该重组能抑制IP3受体的表达,从而反过来作用于TCR介导的信号通路。
B细胞在长期的激素治疗中,激素对B细胞的主要作用在于减少脾脏和淋巴结内的B细胞数量,减少早期B细胞祖细胞的增殖,从而减少IgG并增加IgE的产生。低剂量的激素治疗似乎对免疫球蛋白的合成无明显影响,然而高剂量的激素能增加免疫球蛋白的分解代谢,从而减少其合成,最终降低循环抗体水平[14]。循环B细胞激活因子 (BAFF)是肿瘤坏死因子家族的一员,能够调节B细胞存活、成熟、抗体产生和免疫球蛋白转换。而且BAFF能作为共刺激因子激活T细胞。高剂量的地塞米松(40 mg/d×5d)能在mRNA水平和蛋白水平显著减少BAFF的量[15],从而调控B细胞的成熟和T细胞的激活。
细胞因子与其他介质糖皮质激素能通过基因调节机制减少炎症基因的转录,包括促炎细胞因子的转录。有明确的证据表明激素抑制IL-1~6,IL-11,IL-16,INF-γ,GM-CSF,TNF-α,MMP-9等。Spiesa等[16]发现用地塞米松预处理后的CD4+T细胞上清中IL-2、TNF-α、INF-γ的量有所减少。
最近,su-PAR被提议为与局灶节段性肾小球硬化(FSGS)发病相关的循环因子。在2/3的FSGS患者中su-PAR水平升高,且移植前高水平的su-PAR预示着移植后FSGS的复发[17]。尽管FSGS动物模型体现su-PAR的重要性,但是目前在临床上还没有su-PAR能导致FSGS发生的证据,因此用现有的方法检测su-PAR水平在临床路径中没有决定性意义。在炎症性肠病和HIV患者中,糖皮质激素的使用能降低患者血清中的suPAR水平[18]。但是肾脏疾病中是否有同样的作用还不可知。
糖皮质激素对足细胞的保护作用
足细胞损伤是肾脏病蛋白尿发生的一个重要环节。足细胞的损伤通常包括去分化、裂孔膜或足细胞骨架的直接损伤、肾小球基膜和足细胞间相互作用的变化[19]。人足细胞表达糖皮质激素受体复合物(GR、HSP90、FKBP51、FKBP52),且能与地塞米松结合并转运至细胞核内[20],使糖皮质激素直接保护足细胞成为一种可能。微阵列芯片技术显示地塞米松对足细胞信号通路的基因表达方面有广泛的影响,其中发生显著变化的包括炎症反应、细胞迁移、血管生成、NF-κB和TGF-β通路相关的一些基因[21](图3)。

图3 糖皮质激素的足细胞保护作用
细胞骨架足细胞在维持肾小球通透性方面有着重要作用。在过去的十年里,足细胞被视作蛋白尿性肾脏疾病的始作俑者。我们对足细胞的滤过屏障的认识由静态转为高度动态,因为足突能迅速重组以肌动蛋白为基础的细胞骨架,并且改变其结构与功能。蛋白质组学显示体外培养的分化的足细胞中富含肌动蛋白细胞骨架蛋白、膜联蛋白、应激相关蛋白如热休克蛋白以及抗氧化酶。Ransom等[22]阐述了地塞米松作用于足细胞能使睫状神经因子——一个IL-6样的细胞因子和热休克蛋白27(hsp27)表达增加。Smoyer等[23]在嘌呤霉素诱导的足细胞损伤模型中发现hsp27通过调节肌动蛋白的聚合在调节足细胞形态和肌动蛋白骨架方面起到重要作用。细胞松弛素D、拉春库林A或嘌呤霉素刺激能损伤足细胞,而地塞米松能通过增加聚合的肌动蛋白的总量和肌动蛋白调节相关的GTPase RhoA的活性来维持肌动蛋白丝的稳定性,并且这种保护作用与其他类固醇激素相较而言,是糖皮质激素特异的[24]。同样,地塞米松能通过维持α-actinin-4的表达对阿霉素诱导的足细胞骨架蛋白重排起到保护作用[25]。
细胞凋亡日益增多的证据表明足细胞数量的减少在蛋白尿和肾小球硬化的发生过程中起关键性的作用,包括足细胞从基膜上脱落和凋亡。FSGS动物模型中,泼尼松可增加足细胞祖细胞的数量从而为足细胞提供后备力量,同时减轻肾小球硬化[26]。
细胞凋亡信号通路包括肿瘤抑制蛋白p53,Bcl-2家族和caspase家族。嘌呤霉素通过促进p53介导的凋亡诱导因子(AIF)入核,同时下调磷酸化的胞外信号调节激酶(ERK),诱导足细胞凋亡,而地塞米松能通过降低促凋亡基因p53、Bax的表达,升高抗凋亡基因Bcl-xL的表达和抑制AIF从而抑制嘌呤霉素诱导的足细胞凋亡[27]。有趣的是,当ERK磷酸化被抑制时,地塞米松的凋亡抑制作用也消失,推测地塞米松的足细胞保护作用是ERK磷酸化依赖的。无独有偶,TGF-β也能通过上调Bax及下调Bcl-2的表达造成足细胞损伤,并诱导其凋亡。近期发现microRNA-30家族对维持足细胞骨架和抑制凋亡方面均有重要作用,而地塞米松通过维持足细胞内microRNA-30家族的表达逆转TGF-β对足细胞的促凋亡作用[28]。但是地塞米松不能逆转H2O2造成的足细胞损伤,表明糖皮质激素并没有抗氧化作用,提示可能通过抗氧化应激之外的其他病理机制起到对足细胞的保护作用[27]。
足细胞裂孔膜蛋白Nephrin、podocin、CD2AP和α-actinin-4是重要的足细胞裂孔膜蛋白,在维持裂孔膜的完整性和预防蛋白尿的发生方面起重要作用。
糖皮质激素对足细胞内nephrin蛋白的合成及转录后成熟均有作用。体内试验证实在狼疮性肾炎小鼠,激素能维持足细胞裂孔膜蛋白nephrin和podocin的表达,并改善肾小球组织学改变[29]。Nephrin的磷酸化在维持足细胞的正常结构和功能方面起到重要作用。体外培养的足细胞中,地塞米松一方面能上调nephrin和tubulin-α的表达水平,另一方面能通过调节Nck和Fyn功能从而维持nephrin的磷酸化水平[30]。在阿霉素诱导的非免疫介导的FSGS大鼠模型中,强的松通过稳定nephrin、podocin和CD2AP的表达和亚细胞分布及维持PI3K-Akt-GSK3β信号通路[31]、TRPC6信号通路来保护足细胞[32]。
前景与展望
糖皮质激素通过GR对许多炎症相关基因的表达与调控起到调控作用,并被用于治疗各类炎症性疾病、自身免疫病和肿瘤。临床医师与患者所面临的挑战是细胞、组织器官及个体对糖皮质激素的敏感性不同,临床用药需个体化。GR亚型的发现、与GRE的特异性结合、染色质重塑和共调节因子的招募为糖皮质激素独特的转录调节机制作了很好的解释。糖皮质激素作为免疫抑制剂在临床上的应用是合理及有效的,近年来发现足细胞与免疫细胞共享一些作用靶点,那么糖皮质激素兼顾足细胞的治疗应该更加理想[35]。激素的分子作用机制的面纱正随着研究的深入逐渐被揭开,但是还有很多问题值得去探索,足细胞本身对激素治疗的敏感度与临床治疗的效果是否相关?个体染色质环境差异如何造成激素治疗的差异?是否可以通过联合用药调节GR与共调节因子的相互作用从而增强激素治疗的敏感度?相信随着研究的深入,能进一步从染色质、分子、细胞和个体层面阐述其调节机制,为激素的临床应用提供更合理的理论指导。
1Lu NZ,Collins JB,Grissom SF,et al.Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor.Mol Cell Biol,2007,27(20):7143-7160.
2Ren R,Oakley RH,Cruz-Topete D,et al.Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis.Endocrinology,2012,153(11):5346-5360.
3Hsu SC,DeFranco DB.Selectivity of cell cycle regulation of glucocorticoid receptor function.J Biol Chem,1995,270(7):3359-3364.
4Hudson WH,Youn C,Ortlund EA.The structural basis of direct glucocorticoid-mediated transrepression.Nat Struct Mol Biol,2013,20(1):53-58.
5John S,Sabo PJ,Thurman RE,et al.Chromatin accessibility pre-determines glucocorticoid receptor binding patterns.Nat Genet,2011,43(3):264-268.
6Ramamoorthy S and Cidlowski JA.Ligand-Induced Repression of the Glucocorticoid Receptor Gene Is Mediated by an NCoR1 Repression Complex Formed by Long-Range Chromatin Interactions with Intragenic Glucocorticoid Response Elements.Mol Cell Biol,2013,33(9):1711-1722.
7Ronacher K,Hadley K,Avenant C,et al.Ligand-selective transactivation and transrepression via the glucocorticoid receptor:role of cofactor interaction.Mol Cell Endocrinol,2009,299(2):219-231.
8Samarasinghe RA,Witchel SF,DeFranco DB.Cooperativity and complementarity Synergies in non-classical and classical glucocorticoid signaling.Cell Cycle,2012,11(15):2819-2827.
9Solito E,Mulla A,Morris JF,et al.Dexamethasone induces rapid serine-phosphorylation and membrane translocation of annexin 1 in a human folliculostellate cell line via a novel nongenomic mechanism involving the glucocorticoid receptor,protein kinase c,phosphatidylinositol 3-kinase,and mitogen-activated protein kinase.Endocrinology,2003,144(4):1164-1174.
10 Wang Y,Wang Y,Feng X,et al.Depletion of CD4(+) T cells aggravates glomerular and interstitial injury in murine adriamycin nephropathy.Kidney Int,2001,59(3):975-984.
11 Wang Y,Wang YP,Tay YC,et al.Role of CD8(+) cells in the progression of murine adriamycin nephropathy.Kidney Int,2001,59(3):941-949.
12 Löwenberg M,Verhaar AP,Bilderbeek J,et al.Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN.EMBO Rep,2006,7(10):1023-1029.
13 Harr MW,Rong Y,Bootman MD,et al.Glucocorticoid-mediated inhibition of Lck modulates the pattern of T cell receptor-induced calcium signals by down-regulating inositol 1,4,5-trisphosphate receptors.J Biol Chem,2009,284(46):31860-31871.
14 Alnemri ES,Fernandes TF,Haldar S,et al.Involvement of BCL-2 in glucocorticoid-induced apoptosis of human pre-B-leukemias.Cancer Res,1992,52(2):491-495.
15 Huard B,Schneider P,Mauri D,et al.T cell costimulation by the TNF ligand BAFF.J Immunol,2001,167(11):6225-6231.
16 Spies CM,Gaber T,Hahne M,et al.Rimexolone inhibits proliferation,cytokine expression and signal transduction of human CD4+ T-cells.Immunol Lett,2010,131(1):24-32.
17 Wei C,El Hindi S,Li J,et al.Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis.Nature Med,2011,17(8):952-960.
18 Kolho KL,Valtonen E,Rintamäki H,et al.Soluble urokinase plasminogen activator receptor suPAR as a marker for inflammation in pediatric inflammatory bowel disease.Scand J Gastroenterol,2012,47(8-9):951-955.
19 Kwoh C,Shannon M B,Miner JH,et al.Pathogenesis of nonimmune glomerulopathies.Annu Rev Pathol,2006,1:349-374.
20 Guess A,Agrawal S,Wei CC,et al.Dose- and time-dependent glucocorticoid receptor signaling in podocytes.Am J Physiol Renal Physiol,2010,299(4):F845-853.
21 Cheng X,Zhao X,Khurana S,et al.Microarray analyses of glucocorticoid and vitamin D3 target genes in differentiating cultured human podocytes.PLoS One,2013,8(4):e60213.
22 Ransom RF,Vega-Warner V,Smoyer WE,et al.Differential proteomic analysis of proteins induced by glucocorticoids in cultured murine podocytes.Kidney Int,2005.67(4):1275-1285.
23 Smoyer WE,Ransom RF.Hsp27 regulates podocyte cytoskeletal changes in an in vitro model of podocyte process retraction.FASEB J,2002,16(3):315-326.
24 Ransom RF,Lam NG,Hallett MA,et al.Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization.Kidney Int,2005,68(6):2473-2483.
25 Liu H,Gao X,Xu H,et al.alpha-Actinin-4 is involved in the process by which dexamethasone protects actin cytoskeleton stabilization from adriamycin-induced podocyte injury.Nephrology (Carlton),2012,17(8):669-675.
26 Zhang J,Pippin JW,Krofft RD,et al.Podocyte repopulation by renal progenitor cells following glucocorticoids treatment in experimental FSGS.Am J Physiol Renal Physiol,2013,304(11):F1375-1389.
27 Wada T,Pippin JW,Marshall CB,et al.Dexamethasone prevents podocyte apoptosis induced by puromycin aminonucleoside:role of p53 and Bcl-2-related family proteins.J Am Soc Nephrol,2005,16(9):2615-2625.
28 Wu J,Zheng C,Fan Y,et al.Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids.J Am Soc Nephrol,2014,25(1):92-104.
29 Moysiadis DK,Perysinaki GS,Bertsias G,et al.Early treatment with glucocorticoids or cyclophosphamide retains the slit diaphragm proteins nephrin and podocin in experimental lupus nephritis.Lupus,2012,21(11):1196-1207.
30 Yu M,Ren Q,Yu SY,Role of nephrin phosphorylation inducted by dexamethasone and angiotensin II in podocytes.Mol Biol Rep,2014,41(6):3591-3595.
31 Yu-Shengyou,Li Y.Dexamethasone inhibits podocyte apoptosis by stabilizing the PI3K/Akt signal pathway.Biomed Res Int,2013,2013:326986.
32 Yu S,Yu L.Dexamethasone Resisted Podocyte Injury via Stabilizing TRPC6 Expression and Distribution.Evid Based Complement Alternat Med,2012,2012:652059.
33 刘志红,足细胞病的治疗:免疫抑制剂,还是足细胞保护.肾脏病与透析肾移植杂志,2010,19(1):1-2.
猜你喜欢
保健医苑(2022年4期)2022-05-05
猪业科学(2022年2期)2022-04-21
现代临床医学(2021年1期)2021-01-26
中国生殖健康(2020年2期)2021-01-18
中华养生保健(2020年8期)2021-01-14
中国生殖健康(2019年8期)2019-01-07
哈尔滨医药(2016年3期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
云南中医学院学报(2015年5期)2015-07-31
医学研究杂志(2015年2期)2015-06-10
