
欧盟医疗器械监管方式对我国的启示
2014-03-21 10:13夏广浩王殊轶上海理工大学医疗器械与食品学院上海200093
中国医疗器械信息 2014年7期
夏广浩 王殊轶 上海理工大学医疗器械与食品学院 (上海 200093)
本文作者曾经服务于德国TÜV 莱茵——欧盟最大的公告机构(Notify Body)之一。作为一线的医疗器械产品审核员,对于欧盟的医疗器械法规体系在操作层面有着深切的认识。本文将从医疗器械法规操作层面研究中欧的差别,并提出适合我国国情的可操作化的医疗器械监管模式。
1.欧盟医疗器械监管的法规体系
欧盟医疗器械监管的法规,主要集中在以下方面:
1.1 欧盟三大医疗器械相关指令
欧盟根据医疗器械产品的特点,制订了三大医疗器械指令对不同器械机器进行针对性地监管。
(1) 《有源植入医疗器械指令》(AIMD, Council Directive 90/385/EEC)
这一指令针对通过电源或其他能源起作用,器械在手术后,全部或部分介入人体,留在体内的产品,如心脏起搏器、体内给药器、除颤器等;
(2) 《医疗器械指令》(MDD, Council Directive 93/42/EEC)
该指令覆盖范围最广,影响面最大,除了有源植入医疗器械和体外诊断器械外,几乎所有的医疗器械都属于该指令的管理范围,包括无源植入物、外科器械、电子器械等;
(3) 《体外诊断医疗器械指令》(IVDD, Council Directive 98/79/EC)
这一指令针对试剂产品、校准物、质控物、仪器、设备或系统等体外诊断医疗器械。[2]
1.2 欧盟医疗器械市场监管的主要职责分工
(1) 成员国主管当局处理警戒系统、决定产品分类、市场监督。
(2) 公告机构职责
执行合格评定程序、授权CE 标识使用、执行监督。
(3) 生产企业的职责
产品分类、选择符合性评定程序、准备技术文件、起草符合性声明、建立并执行警戒系统、符合适用指令要求、建立并维护质量体系。
1.3 上市前管理
欧盟将医疗器械分为四类:I,IIa,IIb 以及III 类。I 类器械,又细分为一类普通,Is(一类灭菌),Im(一类测量)。(注:体外诊断试剂(IVDD),有单独的分类,List A, List B,自测类,其他类。)对于Is,Im,IIa,IIb 以及III 类医疗器械生产企业需要向相应的公告机构(Notified body)提出申请,由公告机构负责审查,审查通过后,公告机构授权生产企业使用有其公告号的CE 标识,此时,产品可以上市流通;对于Is 普通器械,生产企业自行负责产品的质量、安全性和有效性审查,并发表自我符合性声明,并在产品上贴上没有审核公告机构代码的CE 标识,这时,产品可以上市流通。
欧盟给企业根据产品特点及企业的自身需求提供了不同产品符合性路径(见图1 和图2)。
1.4 质量体系要求
欧盟要求生产企业必须按照被批准了的质量体系去研发、生产以及最终检验。生产企业可以对设计和开发控制进行删减,但要确保在符合性声明中反映出对设计和开发控制的删减。[3]对于Is,IIa, IIb 以及III 类医疗器械的生产企业,欧盟要求其质量体系必须得到公告机构的审查,确保其产品能够符合相关指令的要求并建议使用欧盟的协调化标准。[4]欧盟虽然没有强制要求生产企业必须使用欧盟协调化的标准去满足其质量体系的要求,但企业如果不采用欧盟的协调化标准,则需要证明其采用的标准能够满足协调化标准的要求。通常情况下,如果存在协调标准,生产企业都会采用协调标准。
1.5 上市后管理
欧盟医疗器械上市后的管理主要体现在两个方面:
第一,生产企业主动报告。
第二,公告机构在系统上的评价。
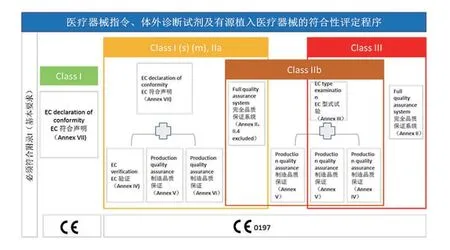
图1. 有源及无源医疗器械
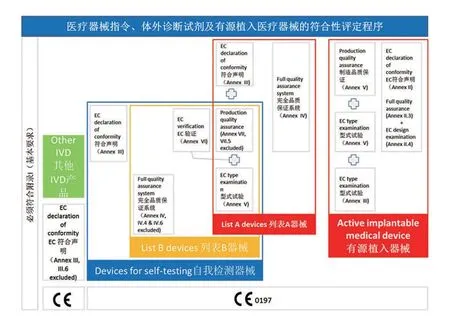
图2. 体外诊断试剂及有源植入医疗器械
2.欧盟与我国医疗器械法规监管体系操作层面的对比
2.1 欧盟法规监管体系的优势
2.1.1 监管实施主体
对于医疗器械的监管,无论是欧盟还是我国,都分为器械上市前监管以及上市后的监管。
(1) 器械上市前的监管
欧盟对于不同的产品类型,有不同的监管方式,对于低风险的产品,如I 类普通器械,仅需要简单确认其符合指令要求即可;对于其他类别的器械,欧盟各主管国是通过具有授权的公告机构对生产企业进行审查,必要时,包括对产品设计文档、技术文件以及质量体系进行评估。评估后,当认定所评估的医疗器械符合指令要求时,该医疗器械产品即被准许标识CE 标志,并开始在欧盟市场中流通和使用。[5]从这里,可以了解到,器械入市前的监管主体是公告机构。
我国医疗器械上市前,必须经过国家食品药品监督管理部门的注册审批。[6]
简单地说,欧盟器械上市前监管主体是公告机构,中国则是政府负责。不仅如此,当前我国医疗器械注册存在注册周期冗长与审评工作量大等问题。[7]
(2) 器械上市后的监管
欧盟主管当局,在某种意义上来说,承担了生产企业的顾问作用,在上市后的警戒系统中同时充当与其他主管当局沟通、协调的工作。
然而,公告机构在器械上市后的监管中并不承担关键的角色,仅在评价生产企业的警戒系统程序、审查警戒系统程序的执行情况、评价警戒事件是否影响到企业现有的产品证书时,承担欧盟主管当局与企业的桥梁作用。[8]
生产企业则是该阶段的主体,承担法律责任;对于使用者,欧盟则鼓励其报告不良事件,但并没有法律的约束。
我国的监管,则是政府主导型,无论是从2008 年版本的《医疗器械不良事件监测和再评价管理办法(试行)》还是近期出台生效的《医疗器械质量监督抽查检验管理规定》,都有着深刻地体现。
《医疗器械不良事件监测和再评价管理办法(试行)》中明确规定,国家食品药品监督管理局负责全国医疗器械不良事件监测和再评价工作,组织检查医疗器械生产企业、经营单位和使用单位医疗器械不良事件监测和再评价工作的开展状况,并会同卫生部组织检查医疗卫生机构的医疗器械不良事件监测工作的开展情况;[9]近期出台的《医疗器械质量监督抽查检验管理规定》更是强化了食品药品监督管理部门在器械上市后的监管职责,国家食品药品监督管理部门负责全国抽验工作管理,并组织实施国家抽验工作。地方各级食品药品监督管理部门负责组织实施辖区内的抽验工作。[10]
简言之,欧盟是生产企业主导型,我国是政府主导型。我国香港地区,对于医疗器械的不良事件,目前尚没有正式的呈报制度。[11]
2.1.2 监管的覆盖面
产品上市前的监管,我国与欧盟一样,几乎实现了全覆盖。而上市后,由于监管实施主体的不同,决定其覆盖面的不同。欧盟,由于是生产企业主导,从理论上来说,实现了全面覆盖;我国是政府主导型,由于资源的限制,产品上市后的全面监管具有很大难度。对于不良事件,则由商务部确定并发布医疗器械不良事件重点监测品种;对于产品抽验,则遵循以下几个原则:
第一,对人体有潜在危险,对其安全性、有效性必须严格控制的医疗器械;第二,使用量大、使用范围广,可能造成大面积危害的医疗器械;第三,出现过质量问题的器械;第四,投诉举报较集中的器械;第五,通过医疗器械风险监测发现存在产品质量风险,需要开展质量监督抽验的医疗器械;第六,在既往抽验中被判不符合标准规定的医疗器械[12]。
显而易见,我国对于医疗器械产品上市后监管的覆盖面,尤其是上市后,远远不及欧盟。
2.1.3 监管的成本
上市前,欧盟授权公告机构主导了产品上市的监管,在这个阶段,生产企业根据自身需求,选择合适的公告机构协助产品上市,并支付公告机构相关的费用。欧盟主管当局几乎是没有参与其中(带药医疗器械除外)。因此,欧盟主管当局直接评审的成品几乎为零;而我国,由政府承担了公告机构在这个阶段的责任,因此,我国的监管成本高。
上市后,欧盟监管是生产企业主导型,政府主要是顾问的功能,因此,政府直接监管成本也几乎为零;我国是政府主导型的监管体制,需要人力的投入;与此同时,根据《医疗器械质量监督抽查检验管理规定》第五条:监督抽验的样品获得分为样品购买、样品返还和无偿提供三种方式。[12]由此我们可以看出,政府在样品获得上面还有相应的支出;同时测试成本等相关费用也不可避免。目前,虽然没有官方数据显示我国每年在监管上投入的成本是多少,但定性分析后与欧盟相比,我们很容易得出我国医疗器械法规监管成本远大于欧盟的结论。
2.1.4 监管的效率
从欧盟的监管模式上我们看到,公告机构在医疗器械上市前以及上市后上所起到的作用。欧盟由于公告机构的介入,决定了其监管效率远高于我国。
2.1.5 技术层面的专业程度
一个公告机构,技术层面,通常由产品测试工程师、审核员以及签证官几个核心的角色组成。由于个体的背景不同,决定了工作人员授权范围的不同。并且,这个授权是具有时效性。授权期满,该授权自动失效,相关人员在授权期外不得从事其原先的授权工作。
如何保证授权的持续性呢?公告机构的工作人员必须不断地学习新的技术、法规知识,在授权期满前,向发证委办公室(Certification Office)证明他们的知识、技能是不断更新的,可以满足当前技术和法规要求的,可以继续从事符合性评定工作。产品测试工程师以及审核员大多具有科研单位、研发机构、临床医生或生产企业研发的背景,同时都是相关专业的科班出身。因此,他们各自都具有扎实的理论知识和丰富的实践经验。这些都为公告机构工作人员的技术专业性奠定了坚实的基础。而我国,目前检测机构在很多领域缺乏高级的专业人才,监管队伍专业化程度较低,人员专业素质参差不齐,缺少系统专业培训检测。另外,队伍中人员流动性大,人员的技术搭配不够合理。[12]因此,我国检测机构与公告机构在技术团队的专业度、稳定性方面都有很大的差距。
2.2 欧盟医疗器械法规体系的不足
法国Poly Implant Prothèse(PIP)公司因劣质人工乳房植入物事件,在2010 年被法国政府强制召回,开始进入大众的视野;2011 年,发生一例患者死亡病例,至2011 年11 月23 日法国政府对该产品进行强制召回,全球有近30000 名妇女需要取出有风险的乳房假体。一时间,PIP 公司的公告机构与PIP 的丑闻联系到了一起,并曝光于各大媒体。尽管该公告机构一再强调其工作符合程序要求,而且PIP 公司在其监管过程中有欺骗行为,法国法院还是在2013 年11 月14 日判决该公告机构承担连带责任并开出了500 万欧元的罚单。
让我们深层次地探讨一下,究竟为什么PIP的欺骗行为可以持续,是什么原则造成了这个灾难性的后果。
2.2.1 公告机构与其监管对象之间的契约关系
公告机构的经济来源是生产企业,公告机构本质还是一家商业公司,而商业公司无法避免其逐利的本性。与此同时,生产企业可以自由地选择并更换公告机构。因此,公告机构在履行监管法律责任的同时,需要吸引客户,并取悦客户,这是商业行为。对于公告机构来讲,生产企业既是其监管的对象,又是其需要视为“上帝“的客户。这两种关系的博弈,必然会导致执法不彻底的结果。
2.2.2 法规监管的透明度、公正性无法保障
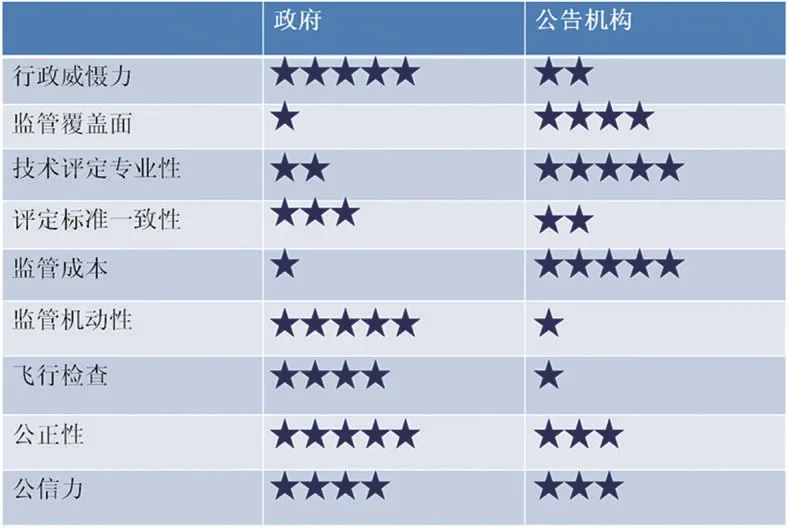
表1. 政府与公告机构监管优势对比
上面的案例中,看似是由于该公告机构的监管不利所造成的,这只是表象,其根本原因在欧盟医疗器械法规监管体制本身。为了加强对生产企业的日常监管,欧盟医疗器械监管的法规体系赋予了公告机构飞行检查的权利,这在理论上是完善的。在实际的监管中,却严重缺乏可操作性。如果公告机构要对生产企业进行检查,无论是认证检查,监督检查,或是飞行检查,公告机构必须要与生产企业进行沟通,确定检查范围、标准、日程以及为此生产企业需要支付的费用等,并签订商业合同。这样的程序,让这个看似美好的机制在操作层面搁浅了。
因此,终究还是法规体制本身的问题。无论多么完美的体制设计,没有考虑到操作性,最终的效果一定会大打折扣。
对以上的分析,总结如下(见表1)。
3.对我国医疗器械法规监管模式的启示
通过以上对于欧盟与我国当前在法规监管操作层面的比较,了解了欧盟与我国在法规监管操作层面的优势与不足,我们可以取长补短,建立一个中国特色的政府主导、第三方唱戏的组合监管模式。要建立这样一种模式,我们必须解决欧盟模式的不足。
(1)解决公告机构与生产企业直接发生契约关系
这是欧盟公告机构工作模式最致命的缺陷。建议企业向政府缴纳相应的上市注册及上市后监管费用,由政府对这些费用进行再分配,切断公告机构与企业的这种契约关系。这样,对公告机构而言,企业的客户身份不再存在,公告机构的客观性、机动性将大大提升。
(2)飞行检查的效果保障
第一个问题解决了,飞行检查的问题也就解决了。如果公告机构在行使任何检查、监管职责之前,不需要与生产企业签订契约,自然可以让飞行检查成为操作很强的上市后监管的有效方式。
(3)解决公正性、公信力的问题
只要打破公告机构与监管企业这种“客户式”监管的模式,从纯技术、纯法规的角度来对医疗器械生产企业履行监管的职责,这两个问题都将得到有效地解决。
(4)如何提升公告机构的行政威慑性
政府可以通过市场化的手段,根据我国的特色,建立一套公告机构的准入和退出制度。政府对公告机构进行适当地授权,让公告机构可以直接影响医疗器械产品的上市和流通。同时,政府需要建立一套对公告机构的监管制度,让其在阳光下行使政府所赋予的监管权,避免公告机构人员利用手中的权利徇私舞弊。另外,政府同样要赋予生产企业申诉或复议的权利,保证生产企业与公告机构在技术层面可以有一个平等的对话平台。这样既解决了公告机构行政威慑性的问题,又保证了生产企业在技术层面与公告机构有一个平等的对话平台。
致谢 :本文在形成过程中,得到了德国TÜV 莱茵大中华区医疗器械产品服务签证官、审核员以及产品测试团队的大力支持和帮助,在此表示感谢。
[1] 岳伟 加快《医疗器械监督管理条例(修正案)》发布的重大意义 中国医疗器械杂志,2013 年37 卷第1 期.
[2] 陈以桢、高惠君 《美国、欧盟医疗器械法规概况及与我国法规的对比》 中国医疗器械杂志,2008 年32 卷第3 期
[3] EUROPEAN COMMITTEE,EN ISO 13485:2012 Medical Devices – Quality management systems – Requirements for regulatory purpose, 2012
[4] THE COUNCIL OF THE EUROPEAN COMMUNITIES, Council Directive 93/42/EEC, 14 June 1996
[5] 曾繁丰、樊翔 《浅谈欧盟医疗器械监管模式》 中国医疗器械信息 2010 年第16 卷第2 期
[6] 岳伟 《我国医疗器械注册制度与美国510K 注册的比较》 中国医疗器械杂志, 2009 年33 卷第1 期
[7] 徐研偌 《美国FDA 最少工作量原则应用给我们的启示》 中国医疗器械信息 2011 年第17 卷第5 期
[8] EUROPEAN COMMITTEE DG Health and Consumers (SANCO), MEDDEV 2.12-1 rev 8 vigilance, January 2013
[9] 国家食品药品监督管理局. 《医疗器械不良事件监测和再评价管理办法(试行)》(国食药监械[2008]766 号). 2008.12.29
[10] 国家食品药品监督管理局. 《医疗器械质量监督抽查检验管理规定》.2013.10.11
[11] 陈欣、宓现强 《香港医疗器械监管概述》 中国医疗器械信息 2010 年第16 卷第7 期
[12] 郑彦云、李丹荣、胡正路 《关于构建在用医疗器械监管体系的几点思考》 中国医疗器械信息 2012 年第10 期
猜你喜欢
护理与康复(2022年6期)2022-11-25
现代仪器与医疗(2022年1期)2022-04-19
生物医学工程学进展(2021年3期)2021-01-20
甘肃教育(2020年18期)2020-10-28
质量安全与检验检测(2019年3期)2019-07-31
质量安全与检验检测(2018年6期)2018-12-28
证券市场红周刊(2018年40期)2018-05-14
证券市场红周刊(2018年41期)2018-05-14
证券市场红周刊(2018年33期)2018-05-14
证券市场红周刊(2018年5期)2018-05-14
