
基于子空间夹角判据监测阿司匹林合成体系中的水杨酸和阿司匹林
2014-03-20 00:40高育新孙阔张祖栋朱淑贞
化学分析计量 2014年3期
高育新,孙阔,张祖栋,朱淑贞
(廊坊市环境监测站,河北廊坊 065000)
阿司匹林又称乙酰水杨酸,其合成过程中水杨酸的准确测定不但可以有效提高阿司匹林生产效率,而且对阿司匹林生产质量的控制有重要意义。2010版《中国药典》[1]以中性乙醇为溶剂、酚酞为指示剂,用NaOH滴定阿司匹林合成体系中的水杨酸和阿司匹林含量,步骤繁琐,误差较大。高效液相法[2–3]测量结果精确度高,但分析时间较长。紫外光谱法作为一种简便快速的分析测试方法,将偏最小二乘法(PLS)应用于分光光度法分析领域,不经分离即可实现多组分的同时测定,但建立PLS训练模型较为繁琐[4]。
专利中利用子空间角判据实现了多组分体系中的组分辨识[5–6],它采用张量空间描述多组分体系的光谱,每一种物质的光谱可用空间中的一个向量表示,同一物质的向量在空间中是相同的,不同光谱特征对应了空间中不同的方向形成向量角度[7]。
笔者以氨基磺酸催化合成阿司匹林体系为研究对象,采用紫外多波长扫描阿司匹林合成过程中样本乙醇溶液的紫外光谱,基于向量–子空间算法模型计算,与单波长线性最小二乘和偏最小二乘结果做比较,实现阿司匹林合成过程的水杨酸和阿司匹林含量分析。
1 实验部分
1.1 主要仪器与试剂
光纤光谱仪:Maya2000Pro型,美国海洋光学公司;
艾科勒分析天平:ALC–210.4型,德国赛多利斯集团;
水杨酸、乙酰水杨酸、乙酸酐、氨基磺酸、无水乙醇:分析纯。
1.2 制备分析模型
1.2.1 背景库和标准样品库
采用1 cm石英比色皿,以无水乙醇为参比,采集样品在250~345 nm波长范围内的紫外光谱。以无水乙醇为溶剂,在2.00~40.00 μg/mL质量浓度范围内,采集质量浓度为2,5,10,15,20,30,40 μg/mL水杨酸标准溶液的紫外光谱;在10.00~200.00 μg/mL范围内,采集浓度为10,20,40,80,120,160,200 μg/mL的阿司匹林标准溶液的紫外光谱;采集乙酸、乙酸酐的紫外光谱。
1.2.2 检验样本的制备
以无水乙醇为溶剂,水杨酸在2.00~40.00 μg/mL质量浓度范围内,阿司匹林在10.00~200.00 μg/mL质量浓度范围内,按均匀实验设计U36(363)和U15(153)建立偏最小二乘校正集和样品检验集并添加适量的氨基磺酸。
1.2.3 阿司匹林的合成与过程监测
按文献方法[8],磁力搅拌下将20.73 g乙酸酐于50 mL三口烧瓶中,水浴温度升至81℃,依次加入13.88 g水杨酸和0.25 g氨基磺酸,恒温反应21 min。每间隔1 min取样100 μL于10 mL容量瓶中并用无水乙醇定容,再取出2 mL以无水乙醇定容于50 mL容量瓶,待测。
1.2.4 数据处理流程
(1)水杨酸定量过程。
步骤1:将被测组分水杨酸的乙醇溶液的系列浓度xi={x1,x2,…,xn}的紫外光谱,经最小二乘法回归后得到多变量回归标准矩阵:yi=aixi+bi,即标准光谱库v。
步骤2:测定不含有水杨酸的系列分析样本阿司匹林、乙酸酐、乙酸、氨基磺酸的乙醇溶液的紫外光谱,并将系列一维谱构成本底谱库N,以矩阵形式存储。
步骤3:测量待测混合物样品乙醇溶液的紫外光谱,作为待定量信号a。
步骤4:从水杨酸光谱库中选择浓度为xi吸收光谱作为被测光谱向量,得到vi。将N,a,vi带入算法中,依据定量精度设定扣减步长Δ,从待测样本的光谱数据的量扣除后的变量记为ak=a–vik/Δ(k=1, 2,…,n),把N和合并后记为Mk,计算Mk与vi的夹角,得到系列夹角θk={θ1,θ2,…,θn}。
步骤5:记录θ最大值θmax所在的k值kmax,待测样品中水杨酸浓度x'=xikmax/Δ。
(2)阿司匹林定量过程。
将上述过程中的水杨酸替换成阿司匹林重复上述步骤,计算阿司匹林样品浓度结果。
以上数据处理及普通最小二乘法(OLS)、PLS均在MATLAB7.8.0中实现。
2 结果与讨论
2.1 各单组分的紫外吸收光谱及背景库建立
在无水乙醇中,水杨酸、乙酸酐、氨基磺酸和乙酰水杨酸在250~345 nm波长范围内的紫外吸收光谱见图1。由图1可知,阿司匹林和水杨酸最大吸收波长分别为275.18 nm和304.21 nm,氨基磺酸在此波长范围内无紫外吸收,水杨酸、阿司匹林、乙酸酐和乙酸4组分光谱有重叠。测定水杨酸浓度时,阿司匹林、乙酸酐和乙酸的乙醇溶液的紫外光谱为背景库;测定阿司匹林浓度时,水杨酸、乙酸酐和乙酸的乙醇溶液的紫外光谱,作为背景库。
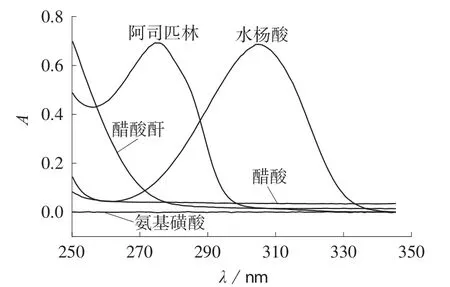
图1 无水乙醇溶液中各单组分紫外光谱
2.2 线性关系的考察及标准库建立
选取波长分别在304.21 nm和275.18 nm波峰点的吸光度值(Y)与水杨酸、阿司匹林标准溶液质量浓度(X)进行最小二乘拟合,建立标准工作曲线,其线性回归方程分别为Y=0.0271X–0.000 8(r=0.999 6),Y=0.005 5X+0.018 8 (r=0.999 3)。结果表明,两组分的质量浓度分别在2.00~40.00 μg/mL和10.00~200.00 μg/mL范围内有良好的线性关系。
把水杨酸标准溶液质量浓度及其对应的全波段的吸光度和阿司匹林准溶液质量浓度及其对应的全波段的吸光度在Matlab计算平台中做最小二乘回归,得到标准光谱库yi=aixi+bi,不同波长下的斜率a和截距b如表1所示。

表1 标准光谱库中各波长下斜率a和截距b
2.3 结果评估
通过检验集15个样本检验模型的定量分析能力,以模型相关系数R、检验均方根差(RMSE)来评价预测效果,同时与OLS、PLS结果做比较。模型的相关参数见表2。
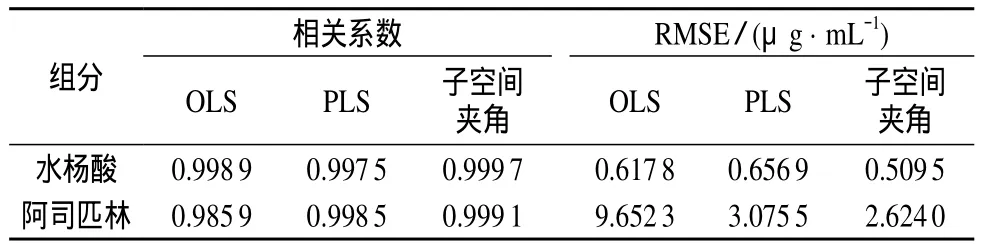
表2 水杨酸和阿司匹林的结果评估参数
如表2所示,304.21 nm波长下阿司匹林、乙酸酐、乙酸、氨基磺酸的紫外吸收对水杨酸的测定无干扰,3种方法对检验集15个样品水杨酸测定结果的相关系数和均方根误差结果均较好;波长在275.18 nm时阿司匹林、水杨酸和乙酸酐均有紫外吸收,水杨酸、乙酸酐的紫外吸收值干扰阿司匹林的测定,选用单波峰点波长的吸光度进行最小二乘线性拟合不能对阿司匹林和水杨酸准确测定。传统OLS方法不能消除强背景干扰,阿司匹林浓度测定结果存在很大的偏差,PLS和基于子空间夹角判定的结果较好,基于子空间夹角判定运算结果优于PLS。
3 模型的应用
3.1 模型对合成体系样品的预测
阿司匹林合成过程中每分钟采集一次样品溶液光谱数据,代入子空间分析模型,结果如表3所示。由表3结果计算反应过程每分钟水杨酸的转化率(如图2所示)。由图2可知,反应温度在81℃,氨基磺酸催化合成阿司匹林的反应在8 min以前反应剧烈,反应体系中反应组分变化较快;8 min后反应较为缓和,此时水杨酸的转化率为97.7%。
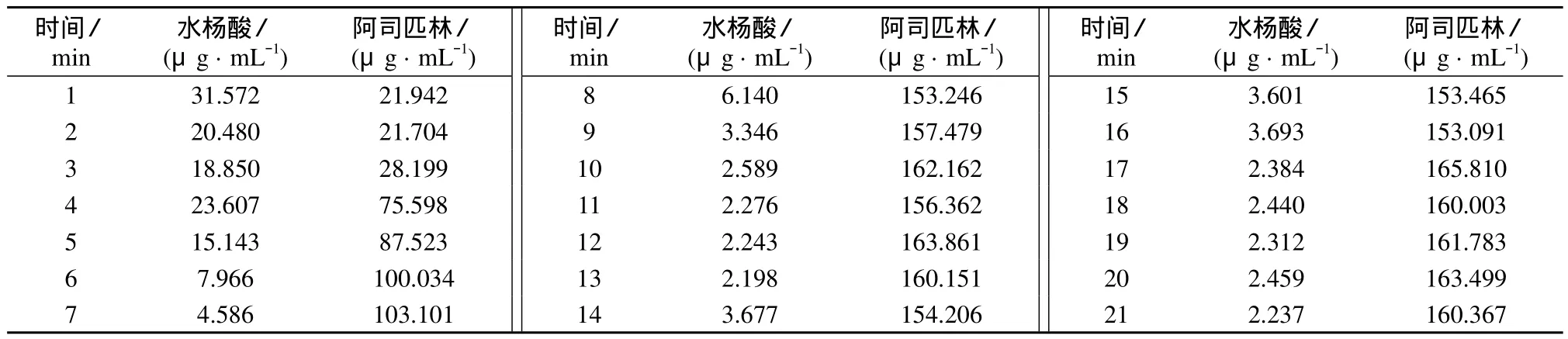
表3 反应过程中样品测定结果
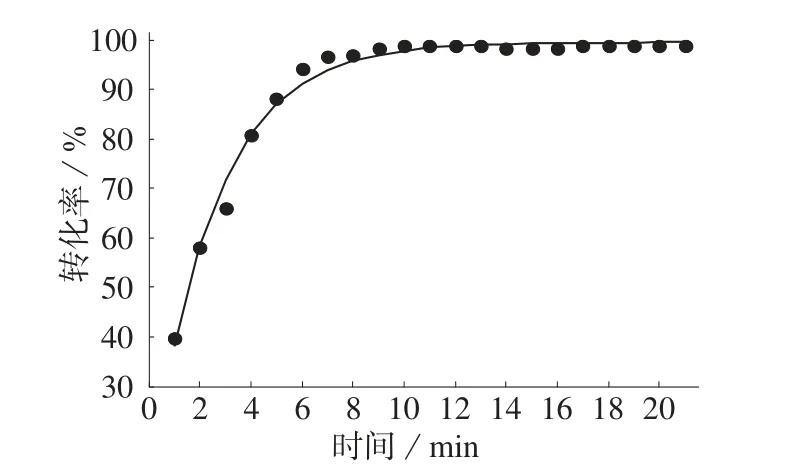
图2 合成过程中水杨酸转化率变化趋势
3.2 加标回收率
选用反应前7 min的7个样品,在标准库样品质量浓度范围内,加入一定量水杨酸和阿司匹林,计算其回收率,结果见表4。由表4可知,阿司匹林和水杨酸的回收率为94.01%~106.44%,相对标准偏差分别为2.85%和3.17%,表明该模型具有良好的准确度与精密度,可满足快速定量分析氨基磺酸催化合成阿司匹林过程体系中的阿司匹林和水杨酸。
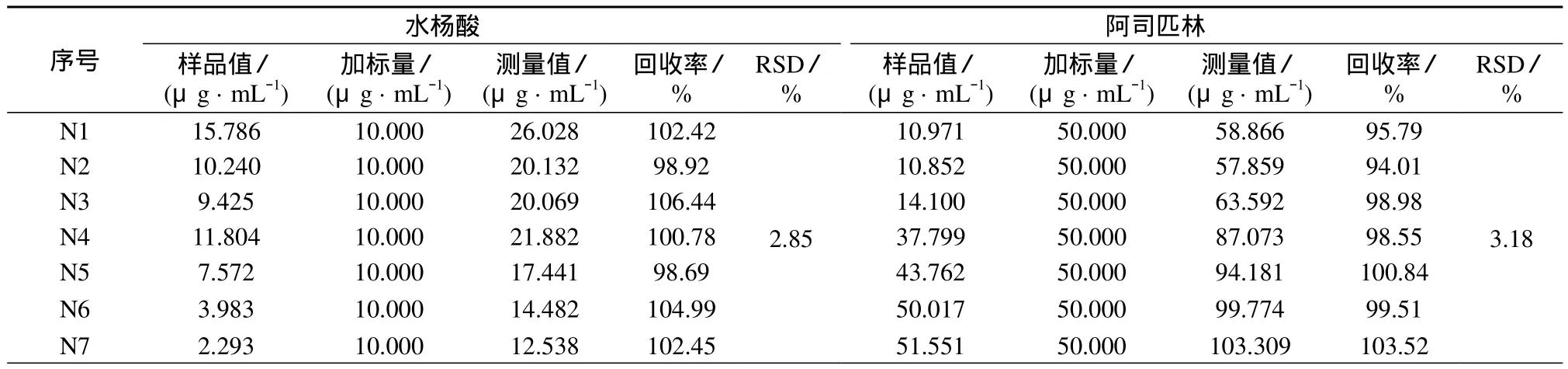
表4 水杨酸和阿司匹林的加标回收率
4 结论
光谱的重叠性影响最小二乘的准确性,传统选用单波峰点的OLS方法不能消除强背景的干扰,PLS和基于子空间夹角判定分析模型均可消除背景光谱的干扰,基于子空间夹角判定无需建立繁琐的校正集进行分析测定。运用子空间夹角模型可准确判定氨基磺酸催化合成阿司匹林过程中水杨酸和阿司匹林的浓度,测定合成样品中水杨酸的浓度和强光谱背景干扰下阿司匹林的浓度,快速判定反应终点。
[1] 国家药典委员会.中华人民共和国药典[M].2部.北京:化学工业出版社,2010: 284.
[2] 李克庆测定阿司匹林肠溶衣片中阿司匹林含量及游离水杨酸[J].中国药师,2006,9(10): 925–927.
[3] Hiral J Panchal,Bhanubhai N Suhagia,Natvarlal J Patel,et al. Simultaneous estimation of atorvastatin calcium,ramipril and aspirin in capsule dosage form by RP–LC[J]. Chromatographia,2009,69: 91–95.
[4] 张小玲,粟晖,姚志湘,等.紫外光谱法监测阿司匹林合成体系中的阿司匹林和水杨酸[J]. 2011,28(5): 1 911–1 915.
[5] 姚志湘,粟晖.基于子空间重合判断的混合光谱模式识别方法:中国,201110188411[P]. 2011–11–30.
[6] 姚志湘,粟晖.基于角度度量的多变量分析方法:中国,201110188187 [P]. 2012–01–04.
[7] 方凤粟晖姚志湘等采用向量扣减方法分析化妆品中对羟基苯甲酸酯[J].分析测试学报,2013,32(6): 732–736.
[8] 杨树.氨基磺酸催化合成乙酰水杨酸的研究[J].昆明师范高等专科学校学报,2007,29(4): 108–109.
猜你喜欢
云南化工(2019年9期)2019-11-11
山东工业技术(2018年11期)2018-06-27
含能材料(2016年10期)2016-05-09
中国药物应用与监测(2015年5期)2015-12-11
化工进展(2015年6期)2015-11-13
中国生化药物杂志(2015年4期)2015-07-07
湖南师范大学自然科学学报(2015年2期)2015-02-27
应用化工(2014年11期)2014-08-16
中国药业(2014年21期)2014-05-26
郑州大学学报(理学版)(2014年4期)2014-03-01
